Clear Sky Science · en
Direct from the seed: an atomic resolution protein structure by ab initio MicroED
Seeing the Smallest Building Blocks of Life
Proteins are the tiny machines that keep living cells running, but to truly understand how they work, scientists need to see their atoms with extraordinary clarity. This article describes how researchers used a powerful electron-based method to determine an atomic-level protein structure starting directly from crushed plant seeds, without relying on prior structural models. Their success offers a new benchmark for studying small or difficult proteins that resist traditional techniques.
Why Tiny Crystals Are a Big Problem
For decades, X-ray crystallography has been the main tool for mapping protein structures at atomic resolution. However, it demands relatively large, highly ordered crystals—often tens to hundreds of micrometers across. Many important proteins naturally form only minuscule crystals, far smaller than this, and therefore slip through the net of standard methods. Another popular approach, single-particle cryo-electron microscopy, struggles with smaller proteins, especially those closer to the typical size of human proteins. Together, these limitations leave a wide swath of biology structurally unexplored.
Using Electrons Instead of X-Rays
Microcrystal electron diffraction, or MicroED, tackles this problem by using electrons instead of X-rays. Electrons interact much more strongly with matter, allowing scientists to extract detailed structural information from crystals thousands of times smaller in volume than those required for X-ray work. In this study, the team focused on crambin, a tiny protein found in the seeds of the oilseed plant Crambe abyssinica. Crambin has long served as a testing ground for advanced crystallographic methods, with earlier work using X-rays and neutrons pushing resolution to astonishing levels. Yet a complete, from-scratch investigation of crambin using MicroED had not been fully realized.
A Fortunate Discovery in a Drying Drop
While purifying crambin from crushed seeds with a simple ethanol-based procedure, the researchers made a striking observation: as a microliter-sized droplet of the solution dried on a slide, it spontaneously produced a dense shower of needle-shaped nanocrystals within seconds. These slivers turned out to be poor performers for X-ray work but nearly ideal for electron diffraction, because they were extremely thin along one dimension and thus transparent to the electron beam. The team also grew traditional, block-like crystals that excelled in X-ray experiments but were too thick for MicroED, even after crushing. This clear reversal—needles bad for X-rays but excellent for electrons, blocks the opposite—highlighted how crystal shape and thickness can dictate which method wins.
Combining Many Needles into One Clear Picture
There was a catch: the needle crystals tended to lie in the same orientation on the microscope grid, meaning that any single crystal provided an incomplete and directionally skewed view of the protein. To overcome this, the researchers adopted a serial strategy, collecting small slices of data from 58 different crystals and merging them into one large dataset. Because the data quality depended strongly on viewing direction, they used a specialized processing step that recognizes and trims away weak, noisy measurements in poorly sampled directions while keeping strong signals where the crystals diffracted best. This treatment produced a very high-quality dataset reaching better than one-ångström resolution, rich enough to reveal individual atoms, including many hydrogens.
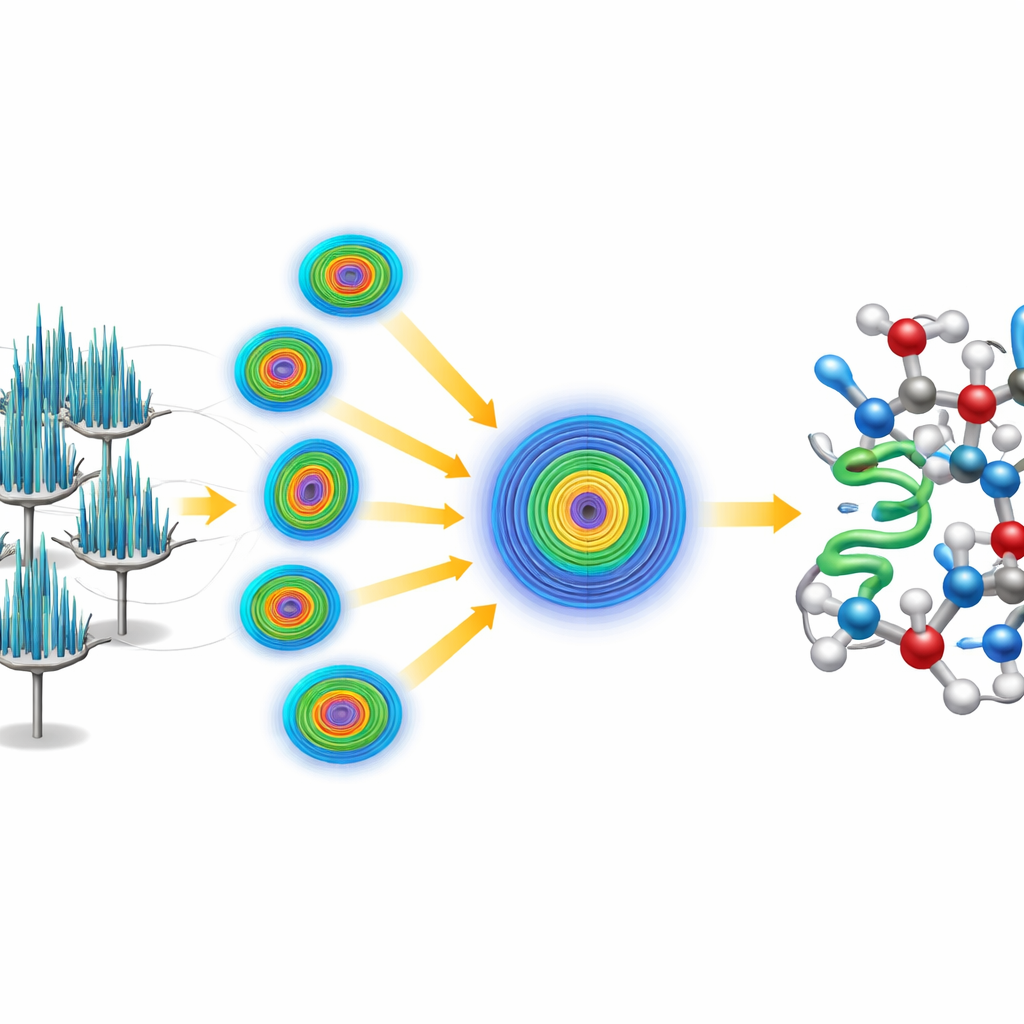
Solving the Structure from Scratch
Instead of leaning on an existing model of crambin, the team challenged themselves to solve the structure “ab initio,” or from first principles. They began with only a tiny, generic five-amino-acid helix—essentially a short, idealized protein fragment unrelated to crambin’s known structure. Using established software, they located where this fragment could plausibly sit within the crystal’s repeating pattern, then applied an iterative sharpening procedure that gradually improved the electron-derived map. The refined map became clear enough for automated software to trace the entire crambin chain without human intervention, yielding a complete, high-resolution model. The data were so detailed that the researchers could distinguish and quantify two naturally occurring variants of crambin that differ at just two positions in the chain.
What This Means for Future Studies
The work shows that, with careful data collection and anisotropy-aware processing, it is possible to reach true atomic resolution and solve a protein structure from scratch using only spontaneously formed nanocrystals and standard electron microscope hardware. Crambin now stands as a public benchmark dataset that other scientists can use to test new analysis methods, refine models of electron scattering, and explore phenomena such as hydrogen bonding and charge distribution. For non-specialists, the key takeaway is that powerful new electron-based tools are extending the reach of structural biology, making it increasingly feasible to visualize the smallest details of proteins that were once considered too small, too fragile, or too unconventional to study in atomic detail.
Citation: Vasireddy, P.C.R., Low-Beer, T., Spoth, K.A. et al. Direct from the seed: an atomic resolution protein structure by ab initio MicroED. Nat Commun 17, 2759 (2026). https://doi.org/10.1038/s41467-026-69601-y
Keywords: MicroED, protein structure, crambin, electron diffraction, nanocrystals