Clear Sky Science · fr
Directement depuis la graine : une structure protéique à résolution atomique par MicroED ab initio
Voir les plus petits éléments constitutifs de la vie
Les protéines sont les minuscules machines qui maintiennent les cellules vivantes en activité, mais pour comprendre vraiment leur fonctionnement, les scientifiques doivent voir leurs atomes avec une clarté extraordinaire. Cet article décrit comment des chercheurs ont utilisé une puissante méthode basée sur des électrons pour déterminer une structure protéique au niveau atomique en partant directement de graines végétales broyées, sans s’appuyer sur des modèles structuraux préexistants. Leur réussite offre un nouveau point de référence pour l’étude de petites protéines ou de protéines difficiles qui résistent aux techniques traditionnelles.
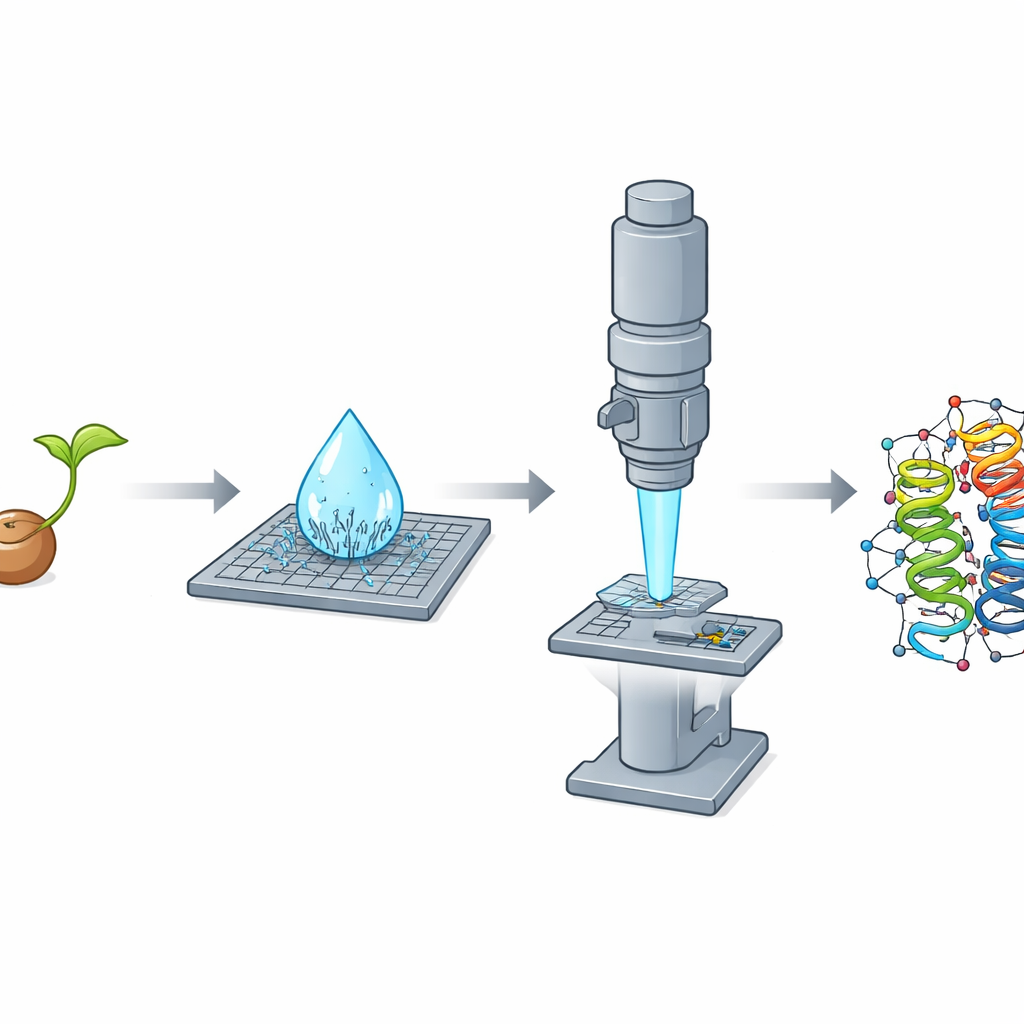
Pourquoi les tout petits cristaux posent un gros problème
Pendant des décennies, la cristallographie aux rayons X a été l’outil principal pour cartographier les structures protéiques à résolution atomique. Toutefois, elle exige des cristaux relativement volumineux et très ordonnés — souvent de l’ordre de dizaines à centaines de micromètres. De nombreuses protéines importantes ne forment naturellement que des cristaux minuscule, bien plus petits, et échappent donc aux méthodes standards. Une autre approche populaire, la cryo-microscopie électronique en particules isolées, peine avec les protéines de petite taille, en particulier celles proches des dimensions typiques des protéines humaines. Ensemble, ces limites laissent une vaste portion de la biologie sans exploration structurale.
Utiliser des électrons au lieu de rayons X
La diffraction électronique sur microcristaux, ou MicroED, s’attaque à ce problème en utilisant des électrons plutôt que des rayons X. Les électrons interagissent beaucoup plus fortement avec la matière, ce qui permet d’extraire des informations structurales détaillées à partir de cristaux dont le volume est des milliers de fois inférieur à celui requis pour la diffraction X. Dans cette étude, l’équipe s’est concentrée sur la crambine, une petite protéine présente dans les graines de la plante oléagineuse Crambe abyssinica. La crambine a longtemps servi de terrain d’essai pour les méthodes cristallographiques avancées, des travaux antérieurs utilisant rayons X et neutrons ayant poussé la résolution à des niveaux étonnants. Pourtant, une investigation complète, « from-scratch », de la crambine par MicroED n’avait pas encore été pleinement réalisée.
Une découverte fortuite dans une goutte qui sèche
Lors de la purification de la crambine à partir de graines broyées par une simple procédure à base d’éthanol, les chercheurs ont fait une observation frappante : lorsqu’une goutte de solution de l’ordre du microlitre séchait sur une lame, elle produisait spontanément en quelques secondes une pluie dense de nanocristaux en forme d’aiguilles. Ces lamelles se sont révélées peu performantes pour les expériences aux rayons X mais presque idéales pour la diffraction électronique, car elles étaient extrêmement fines selon une dimension et donc transparentes au faisceau d’électrons. L’équipe a également fait croître des cristaux traditionnels en blocs qui excellaient en diffraction X mais étaient trop épais pour MicroED, même après concassage. Ce renversement net — aiguilles mauvaises pour les rayons X mais excellentes pour les électrons, blocs l’inverse — a mis en évidence comment la forme et l’épaisseur des cristaux peuvent dicter quelle méthode est la plus adaptée.
Combiner de nombreuses aiguilles pour obtenir une image nette
Il y avait un obstacle : les cristaux en aiguilles avaient tendance à s’allonger dans la même orientation sur la grille du microscope, ce qui faisait qu’un cristal isolé fournissait une vue incomplète et biaisée selon la direction. Pour surmonter cela, les chercheurs ont adopté une stratégie en série, collectant de petites tranches de données sur 58 cristaux différents et les fusionnant en un seul jeu de données. Comme la qualité des données dépendait fortement de la direction de visualisation, ils ont utilisé une étape de traitement spécialisée qui reconnaît et écarte les mesures faibles et bruyantes dans les directions mal échantillonnées tout en conservant les signaux forts là où les cristaux diffractaient le mieux. Ce traitement a produit un jeu de données de très haute qualité atteignant une résolution meilleure que l’ångström, suffisamment riche pour révéler des atomes individuels, y compris de nombreux atomes d’hydrogène.
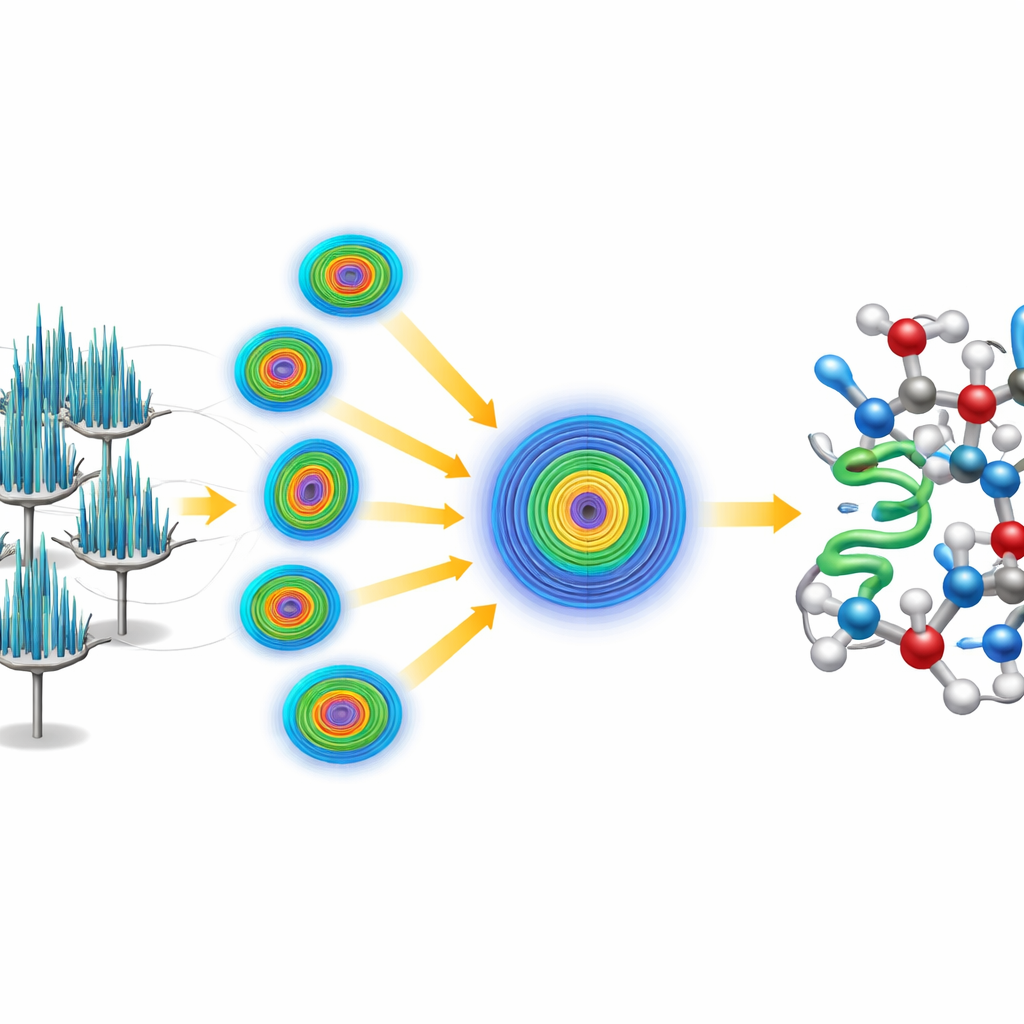
Résoudre la structure à partir de zéro
Plutôt que de s’appuyer sur un modèle existant de la crambine, l’équipe s’est lancé le défi de résoudre la structure « ab initio », c’est‑à‑dire à partir des premiers principes. Ils ont commencé avec un tout petit hélice générique de cinq acides aminés — essentiellement un court fragment protéique idéalisé sans lien avec la structure connue de la crambine. En utilisant des logiciels établis, ils ont localisé où ce fragment pouvait plausiblement se situer dans le motif répétitif du cristal, puis appliqué une procédure itérative d’affinement qui a progressivement amélioré la carte dérivée des électrons. La carte affinée est devenue suffisamment nette pour que des logiciels automatisés puissent tracer l’ensemble de la chaîne de la crambine sans intervention humaine, produisant un modèle complet et à haute résolution. Les données étaient si détaillées que les chercheurs ont pu distinguer et quantifier deux variants naturels de la crambine qui diffèrent à seulement deux positions de la chaîne.
Ce que cela signifie pour les études futures
Ce travail montre qu’avec une collecte de données soigneuse et un traitement prenant en compte l’anisotropie, il est possible d’atteindre une véritable résolution atomique et de résoudre une structure protéique à partir de zéro en n’utilisant que des nanocristaux formés spontanément et du matériel de microscope électronique standard. La crambine devient désormais un jeu de données de référence public que d’autres scientifiques peuvent utiliser pour tester de nouvelles méthodes d’analyse, affiner des modèles de diffusion électronique et explorer des phénomènes tels que les liaisons hydrogène et la distribution de charge. Pour le grand public, la conclusion principale est que de puissants outils électroniques étendent la portée de la biologie structurale, rendant de plus en plus faisable la visualisation des moindres détails de protéines autrefois jugées trop petites, trop fragiles ou trop atypiques pour être étudiées à l’échelle atomique.
Citation: Vasireddy, P.C.R., Low-Beer, T., Spoth, K.A. et al. Direct from the seed: an atomic resolution protein structure by ab initio MicroED. Nat Commun 17, 2759 (2026). https://doi.org/10.1038/s41467-026-69601-y
Mots-clés: MicroED, structure protéique, crambine, diffraction électronique, nanocristaux