Clear Sky Science · fr
Notch-1 dans les macrophages favorise l’ischémie-reperfusion en modulant EZH2/HSF1/BRD4/SIRPα/SHP2 induisant ROS et apoptose dans les cardiomyocytes
Pourquoi la réparation du cœur peut parfois l’endommager
Lorsque les médecins rouvrent une artère coronarienne bouchée après un infarctus, le sang revient brusquement dans un tissu affamé d’oxygène. Cette révascularisation, vitale, peut paradoxalement aggraver les lésions cardiaques. Cette étude examine pourquoi les propres cellules immunitaires de l’organisme, censées aider à la réparation, peuvent au contraire empirer cette « lésion de reperfusion » et met en lumière des interrupteurs moléculaires susceptibles d’être ciblés pour protéger le cœur dans de futurs traitements.
La lutte interne dans un cœur en réparation
Après le rétablissement du flux sanguin, les cellules musculaires cardiaques subissent une tempête de contraintes : variations d’oxygène soudaines, salves de molécules instables et vagues d’inflammation. Les macrophages — cellules immunitaires de première ligne qui affluent dans le tissu lésé — jouent un rôle central dans l’orchestration de la suite des événements. Selon leur mode d’activation, ils peuvent soit aider au nettoyage et à la réparation, soit favoriser l’œdème et la mort cellulaire. Les chercheurs se sont focalisés sur un système de signalisation au sein des macrophages appelé Notch‑1, pour savoir s’il incline l’équilibre vers la guérison ou vers le dommage lors de l’ischémie‑reperfusion.
Approche par gènes, rats et cellules cardiaques
L’équipe a d’abord exploité un jeu de données publics d’expression génique provenant de patients ayant subi un infarctus aigu. Ils ont identifié plus de 100 gènes dont l’activité variait nettement, notamment dans des voies liées à l’inflammation et à la mort cellulaire programmée. Des gènes associés à Notch se sont distingués, suggérant que cette voie était active lors des lésions cardiaques. Les scientifiques sont ensuite passés à des modèles chez le rat, obstruant puis rouvrant temporairement une artère cardiaque pour reproduire la situation clinique. Ils ont injecté aux animaux des macrophages modifiés pour renforcer ou altérer la signalisation Notch‑1 et ont évalué la fonction de pompage cardiaque par échographie, la structure tissulaire au microscope, ainsi que l’étendue des zones nécrotiques et des cellules en détresse.
Comment un interrupteur immunitaire cause des dégâts
Les rats recevant des macrophages présentant une activité élevée de Notch‑1 ont eu des résultats plus mauvais : leur cœur pompait moins efficacement, présentait une désorganisation structurelle accrue et contenait des zones mortes plus étendues ainsi qu’un plus grand nombre de cellules en voie d’apoptose. En modulant soigneusement d’autres molécules, l’équipe a cartographié une chaîne d’événements en aval de Notch‑1 dans les macrophages. Dans cette cascade, Notch‑1 réprimait un régulateur génique appelé EZH2, ce qui libérait d’autres protéines — notamment BRD4, HSF1, SIRPα et SHP2 — à s’activer davantage. Ensemble, ces molécules poussaient les macrophages vers un état fortement inflammatoire favorisant la surproduction d’espèces réactives de l’oxygène, formes d’oxygène chimiquement agressives susceptibles de perforer les membranes cellulaires et d’endommager l’ADN.
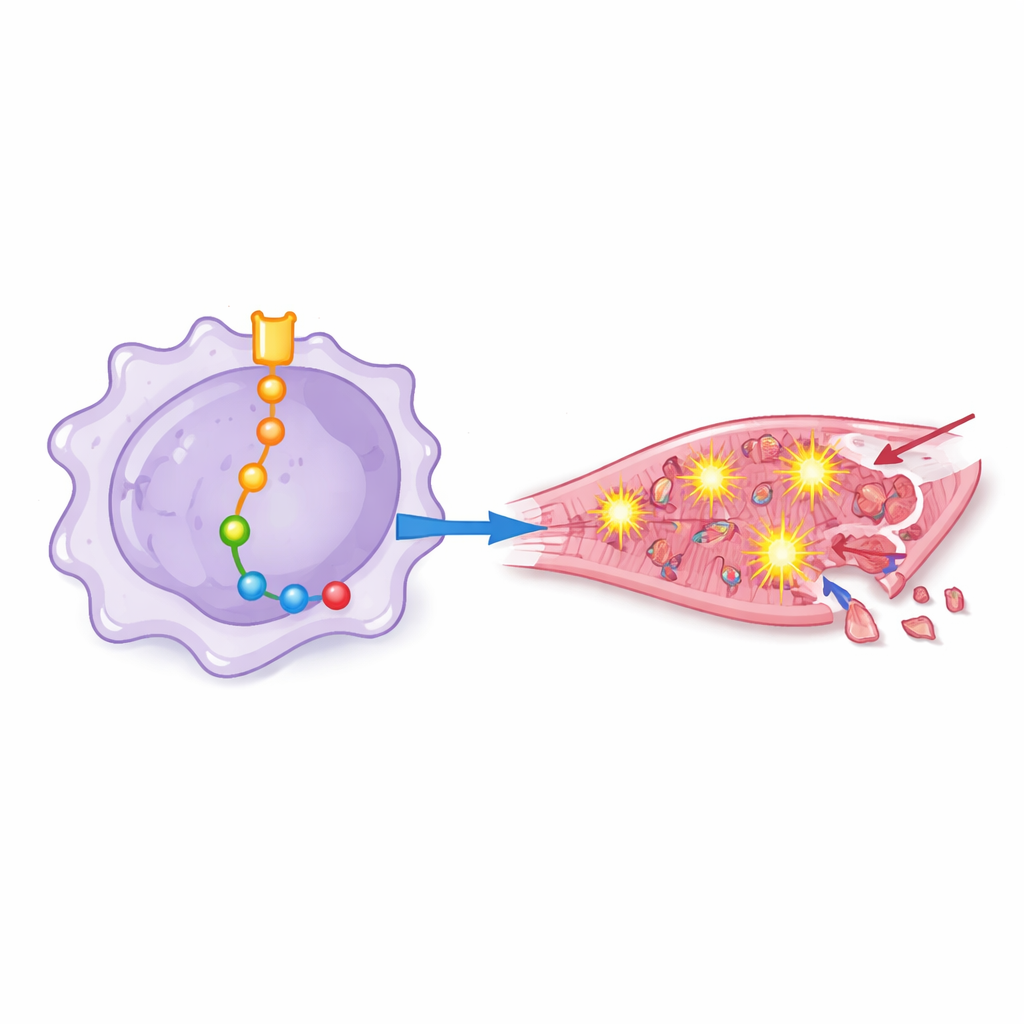
Du stress chimique à la mort des cellules cardiaques
Pour évaluer l’effet de ces macrophages « amorcés » sur des cellules cardiaques humaines, les chercheurs ont exposé des cardiomyocytes en culture au milieu prélevé sur des cultures de macrophages différemment modifiées. Le liquide provenant de macrophages à signalisation Notch‑1 élevée a déclenché un stress oxydatif accru, une activation plus importante d’un complexe inflammatoire connu sous le nom d’inflammasome et une hausse des marqueurs de mort cellulaire programmée. Lorsque Notch‑1 ou certaines protéines en aval étaient inhibées, les cellules cardiaques présentaient moins de signes de dommages oxydatifs, maintenaient de meilleures défenses antioxydantes et avaient moins de risque de mourir. Cela montre que les effets délétères ne sont pas de simples changements moléculaires abstraits mais se traduisent directement par une perte de cellules cardiaques.
Ce que cela implique pour les soins cardiaques futurs
En termes simples, l’étude révèle qu’un interrupteur de signalisation appelé Notch‑1 au sein des macrophages peut les orienter vers un mode nocif pendant la fenêtre critique du rétablissement du flux sanguin vers le cœur. Par une chaîne moléculaire à plusieurs étapes impliquant EZH2, HSF1, BRD4, SIRPα et SHP2, ces cellules inondent le muscle cardiaque adjacent de molécules d’oxygène destructrices et de signaux déclenchant l’apoptose, agrandissant ainsi la lésion. Bien que les travaux aient été menés in vitro et chez l’animal, ils mettent en avant de nouvelles cibles pour des médicaments visant à calmer des macrophages hyperactifs et limiter les dommages collatéraux lors de la réouverture d’une artère obstruée — ce qui pourrait aider à préserver davantage de tissu cardiaque après un infarctus.
Citation: Tong, C., Zhang, J., Zuo, Y. et al. Notch-1 in macrophages promoted the ischemia-reperfusion via modulating EZH2/HSF1/BRD4/SIRPα/SHP2 induced ROS and apoptosis in cardiomyocyte. Sci Rep 16, 11020 (2026). https://doi.org/10.1038/s41598-026-40683-4
Mots-clés: ischémie-reperfusion myocardique, macrophages, signalisation Notch-1, stress oxydatif, apoptose des cardiomyocytes