Clear Sky Science · de
Notch-1 in Makrophagen förderte die Ischämie‑Reperfusionsverletzung über die Modulation von EZH2/HSF1/BRD4/SIRPα/SHP2‑induziertem ROS und Apoptose in Kardiomyozyten
Warum Herzwiederherstellung manchmal dem Herzen schadet
Wenn Ärzte eine verstopfte Herzarterie nach einem Herzinfarkt wieder eröffnen, strömt Blut in das zuvor unterversorgte Gewebe zurück. Diese Rettung, Reperfusion genannt, kann Leben retten – gleichzeitig kann sie das Herz jedoch weiter schädigen. Diese Studie untersucht, warum körpereigene Immunzellen, die eigentlich bei der Heilung helfen sollen, stattdessen diese „Reperfusionsverletzung“ verschlimmern können, und benennt molekulare Schalter, die in zukünftigen Therapien zum Schutz des Herzens verändert werden könnten.
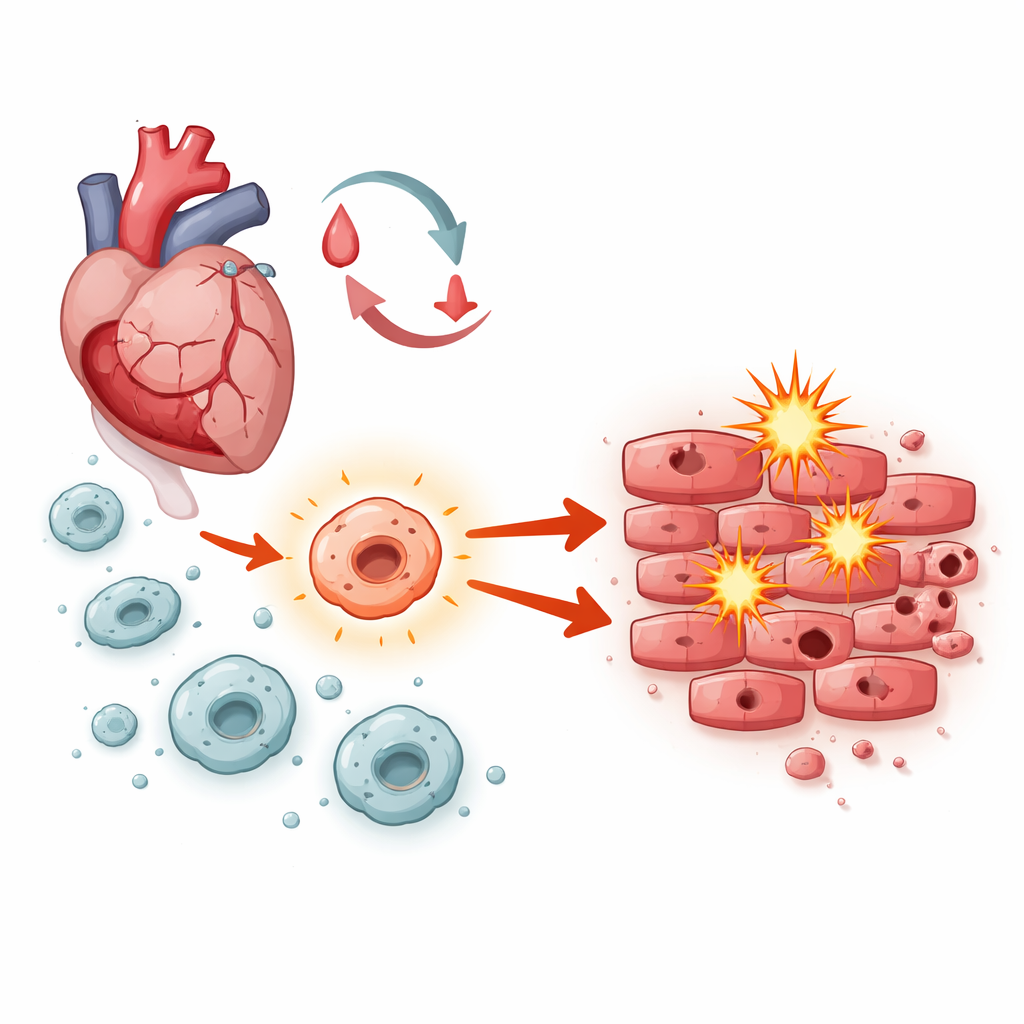
Der innere Wettstreit im heilenden Herzen
Nachdem der Blutfluss wiederhergestellt ist, sind Herzmuskelzellen einem Sturm von Stressfaktoren ausgesetzt: plötzliche Sauerstoffschwankungen, Ausbrüche instabiler Moleküle und Wellen der Entzündung. Makrophagen – die vordersten Immunzellen, die in geschädigtes Gewebe einwandern – spielen eine zentrale Rolle bei der Koordination des weiteren Verlaufs. Je nachdem, wie sie aktiviert sind, können sie entweder bei der Beseitigung von Trümmern und der Reparatur helfen oder Schwellungen und Zelltod vorantreiben. Die Forschenden konzentrierten sich auf ein Signalnetzwerk in Makrophagen, bekannt als Notch‑1, um zu prüfen, ob es dieses Gleichgewicht während der Ischämie‑Reperfusionsverletzung eher in Richtung Heilung oder in Richtung Schaden kippt.
Gene, Ratten und Herzmuskelzellen unter der Lupe
Das Team wertete zunächst einen öffentlichen Genexpressionsdatensatz von Menschen aus, die einen akuten Herzinfarkt erlitten hatten. Sie identifizierten mehr als 100 Gene mit deutlich veränderter Aktivität, insbesondere in Pfaden, die mit Entzündung und Zelltod verbunden sind. Notch‑verwandte Gene fielen dabei besonders auf, was darauf hindeutete, dass dieser Weg während der Herzschädigung aktiviert ist. Anschließend nutzten die Wissenschaftler Ratten, bei denen sie vorübergehend eine Herzarterie verschlossen und wieder eröffneten, um das Patientenereignis zu simulieren. Sie transfundierten den Tieren Makrophagen, die so verändert waren, dass Notch‑1‑Signale verstärkt oder verändert wurden, und untersuchten die Herzpumpfunktion per Ultraschall, die Gewebestruktur unter dem Mikroskop sowie das Ausmaß von Nekrose und sterbenden Zellen.
Wie ein Immun‑Schalter Schaden anrichtet
Ratten, die Makrophagen mit hoher Notch‑1‑Aktivität erhielten, schnitten schlechter ab: Ihre Herzen pumpten weniger effizient, zeigten mehr strukturelle Desorganisation und größere Areale abgestorbenen Muskelgewebes sowie mehr sterbende Zellen. Durch gezieltes Hinzufügen oder Blockieren weiterer Moleküle kartierten die Forschenden eine Kaskade von Ereignissen stromabwärts von Notch‑1 in Makrophagen. In dieser Kette dämpfte Notch‑1 einen Genregulator namens EZH2, was wiederum andere Proteine – darunter BRD4, HSF1, SIRPα und SHP2 – befähigte, aktiver zu werden. Gemeinsam trieben diese Moleküle Makrophagen in einen stark entzündlichen Zustand, der die Überproduktion reaktiver Sauerstoffspezies förderte – die chemisch aggressiven Sauerstoffformen, die Zellmembranen durchlöchern und DNA schädigen können.
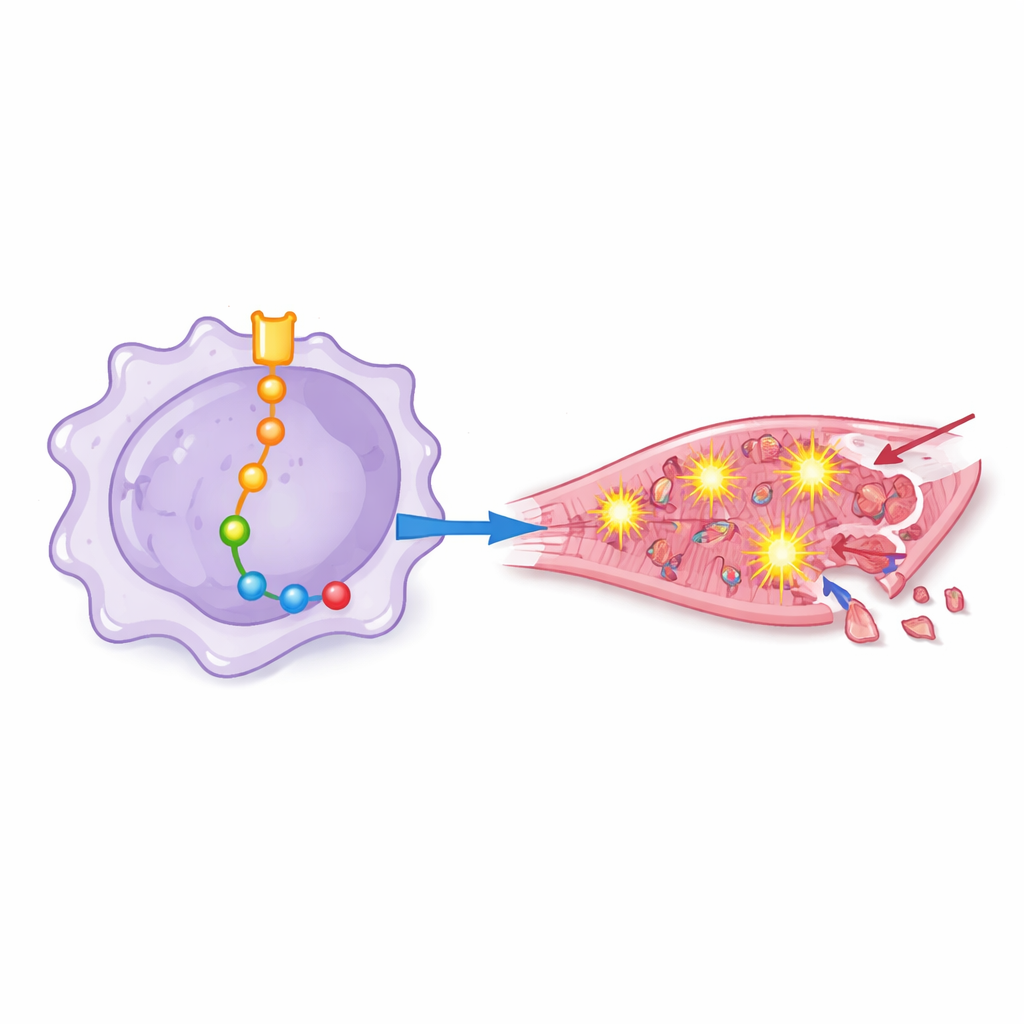
Vom chemischen Stress zum Absterben von Herzmuskelzellen
Um zu prüfen, wie diese „vorkonditionierten“ Makrophagen menschliche Herzmuskelzellen beeinflussen, tränkten die Forschenden kultivierte Kardiomyozyten mit Flüssigkeit aus unterschiedlichen Makrophagenkulturen. Das Medium von Makrophagen mit starker Notch‑1‑Signalgebung löste höheren oxidativen Stress, stärkere Aktivierung des entzündlichen Proteinkomplexes (Inflammasom) und einen Anstieg von Markern für programmierten Zelltod aus. Wurden Notch‑1 oder bestimmte nachgeschaltete Proteine blockiert, zeigten die Herzzellen weniger Anzeichen oxidativer Schäden, behielten bessere antioxidative Abwehrmechanismen und starben seltener. Dies belegt, dass die schädlichen Effekte nicht nur abstrakte molekulare Veränderungen sind, sondern direkt in Herzzellverlust übersetzt werden.
Was das für die künftige Herzmedizin bedeutet
Vereinfacht gesagt zeigt die Studie, dass ein Signalschalter namens Notch‑1 in Makrophagen sie in dem kritischen Zeitfenster nach Wiedereröffnen des Blutflusses in einen schädlichen Modus drängen kann. Über eine mehrstufige molekulare Kaskade, an der EZH2, HSF1, BRD4, SIRPα und SHP2 beteiligt sind, überfluten diese Zellen das umliegende Herzmuskelgewebe mit zerstörerischen Sauerstoffmolekülen und Signalen, die Zelltod auslösen und die Verletzung vergrößern. Zwar wurden die Arbeiten an Zellen und Tieren durchgeführt, doch sie heben neue Zielstrukturen für Medikamente hervor, die überaktive Makrophagen dämpfen und Kollateralschäden beim Wiedereröffnen einer verschlossenen Arterie begrenzen könnten – womit in Zukunft nach einem Herzinfarkt mehr Herzmuskelgewebe gerettet werden könnte.
Zitation: Tong, C., Zhang, J., Zuo, Y. et al. Notch-1 in macrophages promoted the ischemia-reperfusion via modulating EZH2/HSF1/BRD4/SIRPα/SHP2 induced ROS and apoptosis in cardiomyocyte. Sci Rep 16, 11020 (2026). https://doi.org/10.1038/s41598-026-40683-4
Schlüsselwörter: myokardiale Ischämie‑Reperfusion, Makrophagen, Notch‑1‑Signalweg, oxidativer Stress, Apoptose von Kardiomyozyten