Clear Sky Science · fr
TIGAR régule l’intégrité de la barrière de mucus intestinale en inhibant la lactylation de G6PD/6PGD dans la rectocolite hémorragique
Pourquoi la couche visqueuse de l’intestin compte
L’épithélium intestinal est recouvert d’une couche de mucus glissante qui maintient des trillions de microbes à distance. Dans la rectocolite hémorragique, une maladie chronique qui enflamme le côlon, ce manteau protecteur s’amincit ou se fissure souvent, laissant les bactéries et les irritants atteindre la paroi intestinale. Cette étude explore pourquoi cette barrière de mucus défaillit, retraçant le mécanisme depuis le métabolisme du sucre au sein de cellules spécialisées sécrétant le mucus jusqu’aux lésions intestinales — et met en lumière une molécule, TIGAR, comme une cible thérapeutique prometteuse.
Un défenseur discret dans les cellules intestinales
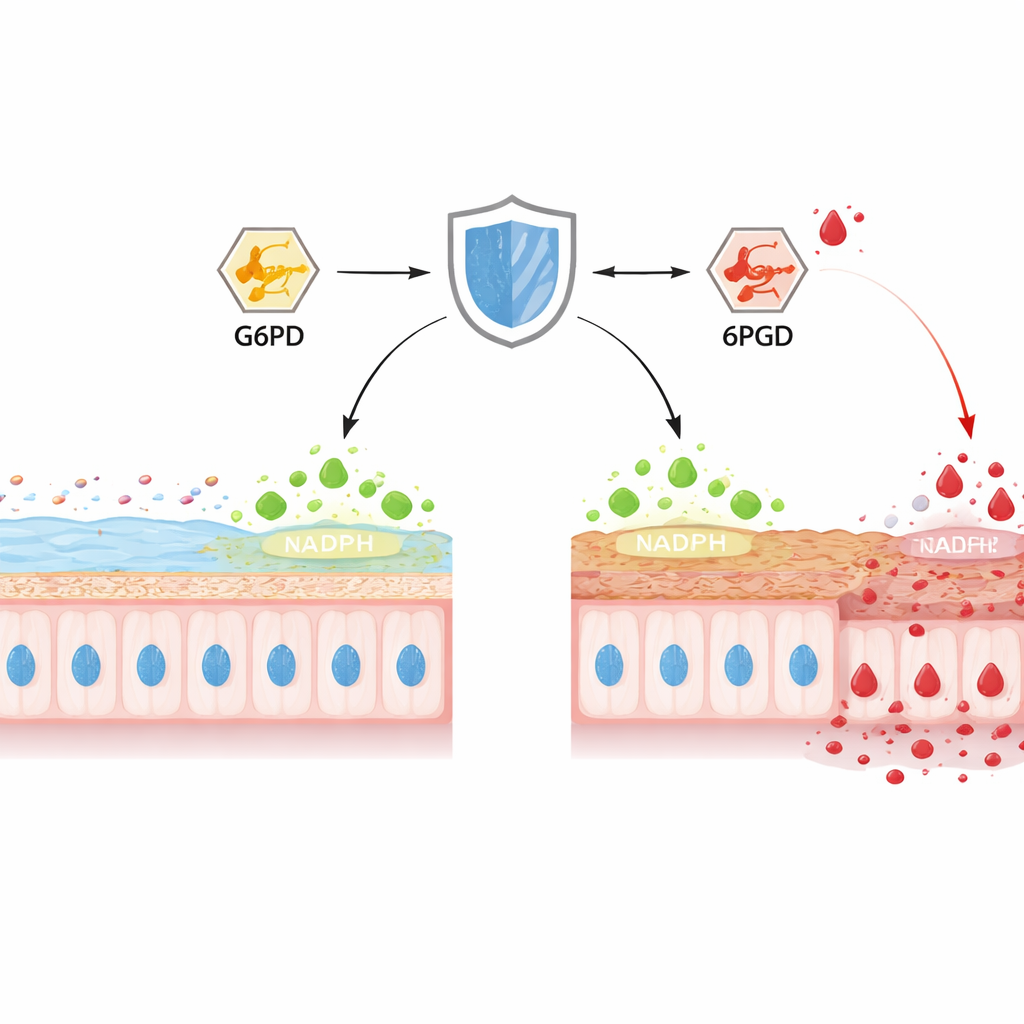
Dans le côlon, les cellules caliciformes fabriquent en continu la MUC2, la protéine principale du mucus qui forme le gel protecteur interne. Les auteurs se sont concentrés sur TIGAR, une protéine connue pour orienter le glucose vers une voie produisant du NADPH, cette « puissance réductrice » essentielle pour neutraliser les oxydants nocifs. Chez la souris soumise à une colite chimique, les niveaux de TIGAR dans le côlon ont chuté sur plusieurs jours, au même rythme que l’amincissement de la couche de mucus, l’érosion de la surface épithéliale et la progression des bactéries vers les tissus. Lorsque les chercheurs ont supprimé sélectivement TIGAR uniquement dans l’épithélium intestinal des souris, la colite est apparue plus tôt et est devenue plus sévère, avec des cryptes déformées, moins de cellules caliciformes et une MUC2 nettement moins mature. Des mesures biochimiques ont révélé un épuisement du NADPH, un affaiblissement des systèmes antioxydants et des pics d’espèces réactives de l’oxygène et de l’azote, liant la perte de TIGAR à un stress oxydatif intense et à l’effondrement de la barrière.
Un usage du sucre réorienté vers une combustion improductive
Pour comprendre comment TIGAR façonne le métabolisme, l’équipe a profilé des centaines de petits métabolites dans des lignées cellulaires apparentées aux cellules caliciformes et dans des tissus coliques de souris. La suppression de TIGAR n’a pas modifié la quantité de glucose importée par les cellules ni la consommation globale d’oxygène de l’animal, mais elle a profondément changé le devenir de ce glucose. En utilisant du sucre marqué, ils ont montré qu’un moindre flux de carbone transitait par la voie des pentoses phosphates — principale source de NADPH — tandis qu’une plus grande part était poussée vers la glycolyse classique, aboutissant au lactate. Deux métabolites se sont distingués : le 6‑phosphogluconate, un carrefour de la voie des pentoses phosphates, s’accumulait fortement, et les niveaux de lactate grimpaient dans les cellules et les tissus dépourvus de TIGAR, en particulier lors de la colite. Les données d’expression génique laissaient entendre une stimulation des enzymes de la glycolyse, alors que les niveaux protéiques de deux enzymes clés de la voie des pentoses phosphates, G6PD et 6PGD, restaient inchangés, suggérant que leur activité — et non leur abondance — était inhibée.
Une marque chimique qui neutralise les enzymes antioxydantes
Les chercheurs ont alors mis au jour le mécanisme d’inhibition : la lactylation, une marque chimique ajoutée aux protéines en utilisant le lactate comme donneur. Dans les modèles de colite et dans des cultures cellulaires inflammatoires, la lactylation protéique globale augmentait, et G6PD ainsi que 6PGD portaient des niveaux élevés de cette modification. La spectrométrie de masse a identifié des positions lysine spécifiques sur chaque enzyme — K432 sur G6PD et K38 sur 6PGD — comme sites majeurs de lactylation. La modélisation structurale et des tests par mutation ont montré que la modification de ces positions gêne la façon dont G6PD forme son dimère actif et la manière dont 6PGD se lie à son cofacteur, réduisant fortement leur capacité à générer du NADPH. Bloquer la production de lactate ou empêcher la lactylation à ces sites a restauré l’activité enzymatique, augmenté le NADPH, réduit le stress oxydatif et amélioré la maturation de la MUC2. Chez les souris déficientes en TIGAR, l’administration de versions mutantes de G6PD et 6PGD incapables d’être lactylées a préservé l’épaisseur du mucus et réduit l’intrusion bactérienne dans la couche interne.
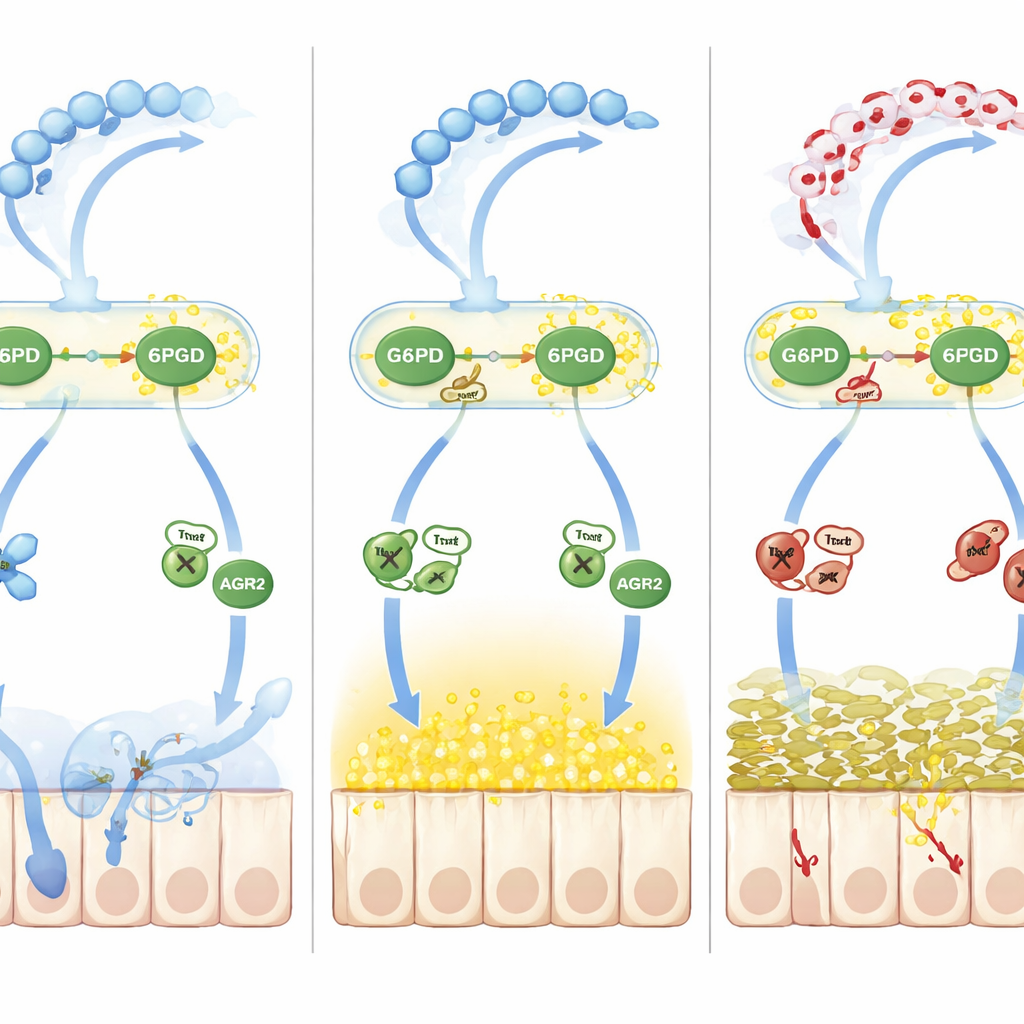
Comment le déséquilibre redox perturbe le traitement du mucus
Au‑delà de l’oxydation simple, l’équipe a identifié une deuxième vague de dommages entraînée par le monoxyde d’azote. L’augmentation du monoxyde d’azote dans la colite a accru la S‑nitrosylation, une autre marque chimique réversible portée par les résidus cystéine. Une protéine chaperonne appelée AGR2, essentielle pour convertir les précurseurs de la MUC2 en leur forme mature dans le réticulum endoplasmique, est devenue fortement S‑nitrosylée sur une cystéine critique (Cys81). Ce changement a affaibli l’interaction d’AGR2 avec les précurseurs de la MUC2, provoquant l’accumulation de protéines mal repliées et une augmentation du stress du réticulum endoplasmique, réduisant encore le réservoir de mucus correctement formé. L’intermédiaire clé était la thioredoxine‑1 (Trx1), une enzyme sensible au redox qui peut soit enlever, soit transférer des groupes nitrosyle selon son propre état d’oxydation. Lorsque le NADPH venait à manquer parce que G6PD et 6PGD étaient lactylés et ralenties, Trx1 passait d’un mode dénitrosylant protecteur à un mode transnitrosylant délétère, transférant des groupes nitrosyle sur AGR2 et aggravant le défaut de mucus.
Ce que cela signifie pour les personnes atteintes de colite
Pris ensemble, les résultats décrivent une réaction en chaîne : l’inflammation chronique diminue TIGAR dans les cellules épithéliales intestinales ; cela détourne le glucose des voies produisant le NADPH, augmente le lactate et lactyle G6PD et 6PGD ; la production de NADPH chute, Trx1 devient hyper‑oxydé, AGR2 est excessivement nitrosylée et la MUC2 ne parvient pas à maturer, laissant la barrière de mucus mince et perméable. Pour un lecteur non spécialiste, le message est qu’un léger changement dans la façon dont les cellules intestinales métabolisent le sucre peut se traduire par une atteinte bien concrète du bouclier visqueux de l’intestin. En protégeant la fonction de TIGAR, en empêchant la lactylation délétère de G6PD et 6PGD, ou en rétablissant l’équilibre redox de Trx1 et AGR2, de futurs traitements pourraient renforcer la barrière mucale et contenir l’inflammation dans la rectocolite hémorragique.
Citation: Wu, D., Su, S., Zhang, P. et al. TIGAR regulates intestinal mucus barrier integrity by inhibiting lactylation of G6PD/6PGD in ulcerative colitis. Nat Commun 17, 3382 (2026). https://doi.org/10.1038/s41467-026-70263-z
Mots-clés: rectocolite hémorragique, barrière de mucus intestinale, TIGAR, stress oxydatif, métabolisme du glucose