Clear Sky Science · en
Cysteine-reactive mitigators of small vessel disease-related NOTCH3 mutants
Why this brain blood vessel study matters
Cerebral small vessel disease is a major cause of stroke and dementia, yet many inherited forms have no treatment. One such condition, called CADASIL, is driven by changes in a single protein in the walls of tiny brain arteries, leading to progressive damage over decades. This study explores whether certain small, drug-like chemicals can nudge that faulty protein back toward a healthier shape, hinting at a new way to slow or prevent disease in people who carry risky gene variants.
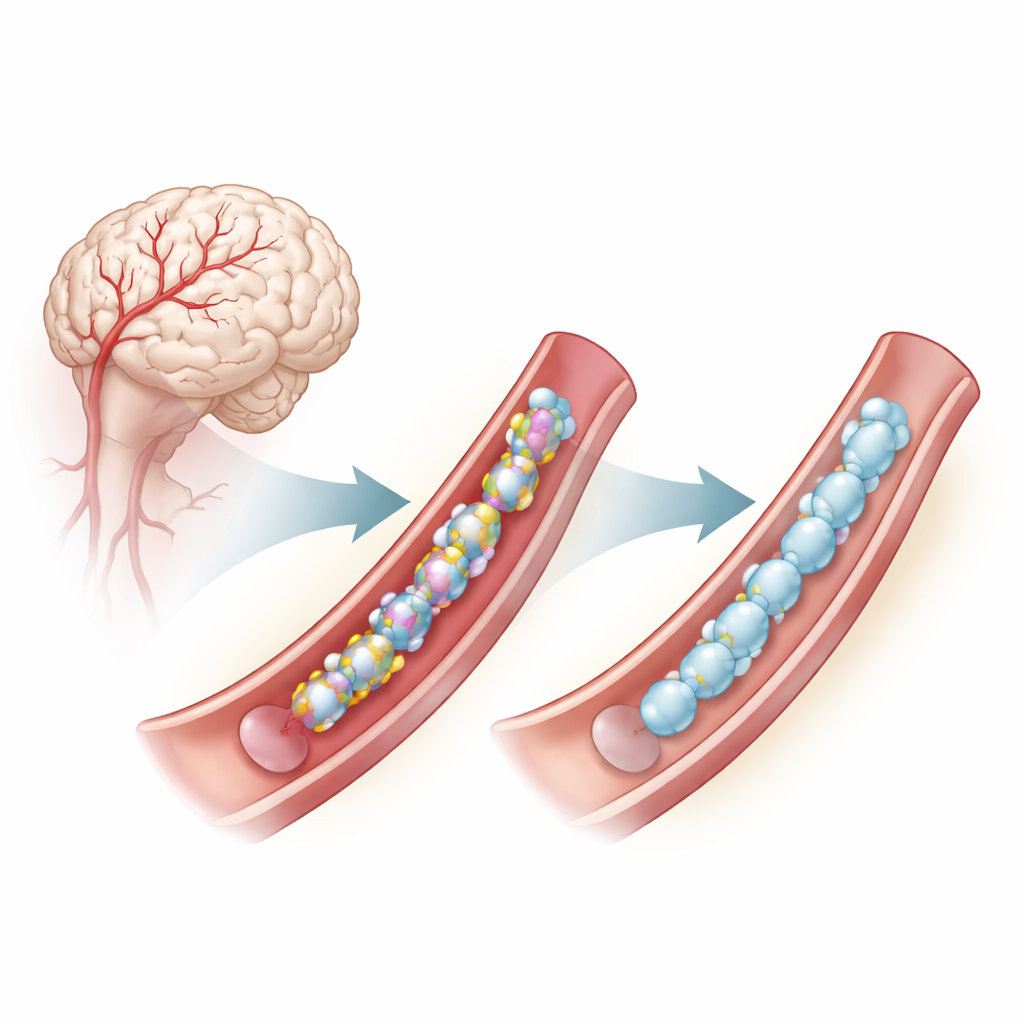
A problem deep inside brain blood vessel walls
CADASIL is caused by mutations in a gene called NOTCH3, which is active in the smooth muscle cells that line small brain arteries. These mutations often alter the number or placement of a particular building block, cysteine, within repeated segments of the NOTCH3 protein. Cysteine units normally pair up to form disulfide bonds, tiny chemical bridges that help the protein fold correctly. When the number of cysteines is wrong, NOTCH3 tends to misfold, clump outside cells, and disrupt blood vessel function, contributing to early strokes and cognitive decline. Even some mutations that do not directly change cysteine appear to create similar misfolding problems dependent on these disulfide bonds.
A light-based test for misshapen proteins
To search for ways to rescue faulty NOTCH3, the researchers used a sensitive laboratory tool they had previously developed: a split-luciferase reporter system called LSL-NOTCH3. In this setup, pieces of the NOTCH3 protein are fused between two halves of a light-emitting enzyme. When NOTCH3 is made, folded, and secreted properly from cells, the enzyme halves rejoin and produce a strong light signal in the surrounding liquid. When the protein misfolds, production and secretion fall, and the light output drops. A second measurement in the same assay tracks how much of the protein has the correct disulfide bonds by comparing light before and after chemically "unlocking" hidden, abnormal bonds. Together, these readouts provide a rapid way to see whether any treatment makes mutant NOTCH3 behave more like its healthy counterpart.
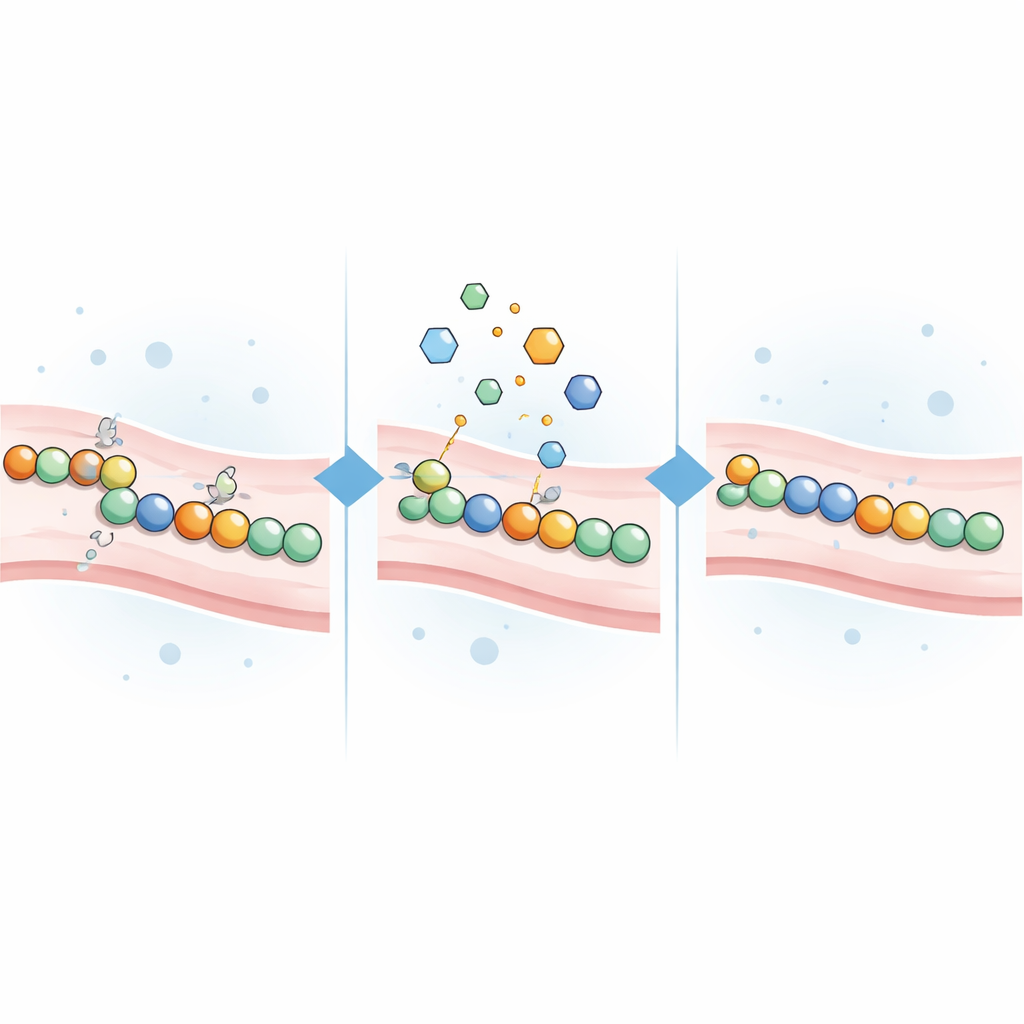
Testing cysteine-seeking chemicals as fixers
Because CADASIL mutations often leave cysteine groups unmatched, the team reasoned that small molecules that react with cysteine might help. They assembled 21 cysteine-reactive compounds, including classic laboratory reagents and several approved or investigational drugs originally developed for cancer, stomach acid disorders, or other conditions. Each compound was added to cells expressing one of 16 different disease-causing NOTCH3 variants spanning several risk-linked regions of the protein, as well as to cells expressing normal or harmless variants. The scientists then measured how much the light signal changed in response to each treatment.
Broad-acting helpers emerge from the screen
More than half of the tested compounds improved at least one disease-causing NOTCH3 variant, and five chemicals boosted the primary light signal in half or more of the mutants. Among these, the strongest and broadest effects came from disulfiram, an established drug for alcohol dependence, and auranofin, a gold-containing drug used for rheumatoid arthritis. These compounds not only increased overall light output, suggesting better protein production and secretion, but also improved the fraction of protein with favorable disulfide bonding patterns. Importantly, they had no effect on normal NOTCH3, implying some selectivity for the misfolded forms. Additional experiments showed that the chemicals needed to be present while cells were making the protein, and did not work on secreted protein alone, indicating that they likely act during folding inside the cell.
Beyond one gene and one mutation
The researchers asked whether the benefit of cysteine-reactive compounds depended on the specific amino acid that replaced cysteine in a mutation. By systematically substituting many different residues at key positions, they found that disulfiram and a related compound still improved reporter activity in nearly all cases. This supports the idea that the drugs are targeting the shared problem of missing cysteine pairs, rather than any particular replacement. They also tested a second, independent reporting system based on a different split-luciferase design and confirmed that disulfiram similarly enhanced secretion of mutant NOTCH3 fragments but not the normal form. When they turned to another cysteine-rich protein involved in Marfan syndrome, FBN1, disulfiram helped only a few mutants and auranofin helped none, suggesting that NOTCH3 in CADASIL may be especially susceptible to this strategy.
What this could mean for future treatments
This work shows that it is possible to partially correct the behavior of many different NOTCH3 mutants using broadly cysteine-reactive small molecules, including drugs already in clinical use for other diseases. The findings point to a unifying mechanism in which extra or exposed cysteine groups in mutant NOTCH3 drive harmful misfolding, and where capping those reactive sites can restore more normal protein folding and secretion. While these results come from cell-based models and not yet from patients or animal studies, they lay the groundwork for developing or repurposing cysteine-targeting drugs as potential therapies for CADASIL and related disorders of small brain vessels.
Citation: Cartee, N.M.P., Zhang, X., Lee, S.J. et al. Cysteine-reactive mitigators of small vessel disease-related NOTCH3 mutants. Sci Rep 16, 14300 (2026). https://doi.org/10.1038/s41598-026-45103-1
Keywords: CADASIL, NOTCH3 mutations, small vessel disease, cysteine-targeting drugs, protein misfolding