Clear Sky Science · en
Recurrent DNA break clusters drive replication-stress-induced copy number variants and genome diversification
Hidden changes in our DNA blueprint
Every cell in our body copies its DNA billions of times over a lifetime, and most of the time this happens flawlessly. But when the copying machinery is strained, pieces of DNA can be lost, duplicated, or rearranged—changes known as copy number variants. These alterations are common in brain disorders and cancers, yet how they arise in the first place has been a long‑standing mystery. This study uncovers a central culprit: special weak spots in long brain genes where DNA breaks tend to pile up, seeding a surprising variety of permanent genetic changes.
Where the genome tends to crack
The researchers focused on “recurrent DNA‑break clusters,” stretches within very long genes in neural progenitor cells—the dividing cells that give rise to neurons. These long genes are heavily used and copied late during the cell’s DNA‑replication schedule, making them especially vulnerable. By gently slowing DNA polymerases with a drug that induces replication stress, the team used deep whole‑genome sequencing to watch where the genome thinned out. They found that small but consistent losses of DNA, and sharper, focal deletions, were strongly concentrated at these break‑prone regions within long neuronal genes, far more often than would be expected by chance.
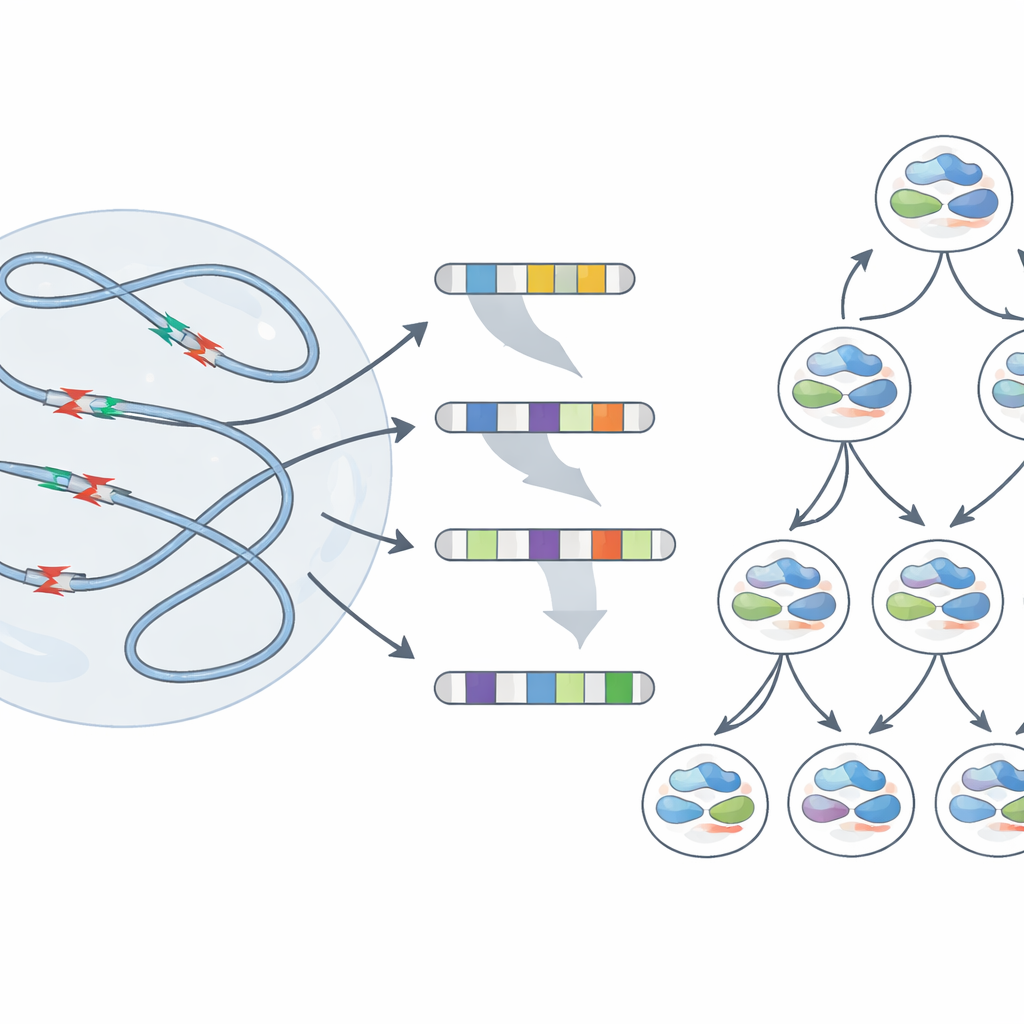
From temporary breaks to lasting scars
Bulk sequencing shows average behavior across millions of cells, but does not reveal what happens in any single cell. To see how individual genomes respond to stress, the team used a single‑cell method that tracks which original DNA strands each daughter cell inherits. After replication stress, many cells carried large deletions extending over millions of DNA letters and even losses of entire chromosome arms. Crucially, the breakpoints of these large changes frequently lined up with the same fragile regions identified earlier. Sometimes, sister cells descending from one stressed ancestor ended up with very different rearrangements—evidence that a single round of damage at these hotspots can set daughter cells off on divergent genetic paths.
Transcription turns fragile spots into mutation factories
The authors then asked what makes these regions so special. They used CRISPR tools to shut down the local on‑switches (promoters and enhancers) that drive transcription of two giant brain genes known to host break clusters. When they turned off transcription, the characteristic break clusters vanished, and the associated deletions no longer appeared, even under replication stress. Notably, the timing of DNA replication at those sites stayed late or even became slightly later, showing that it is not simply “when” in the cell cycle these regions are copied that matters, but the combination of active use and copying. In other words, transcribing long genes while they are being replicated creates conflict zones where breaks form, and those breaks are the seeds of copy number variants.
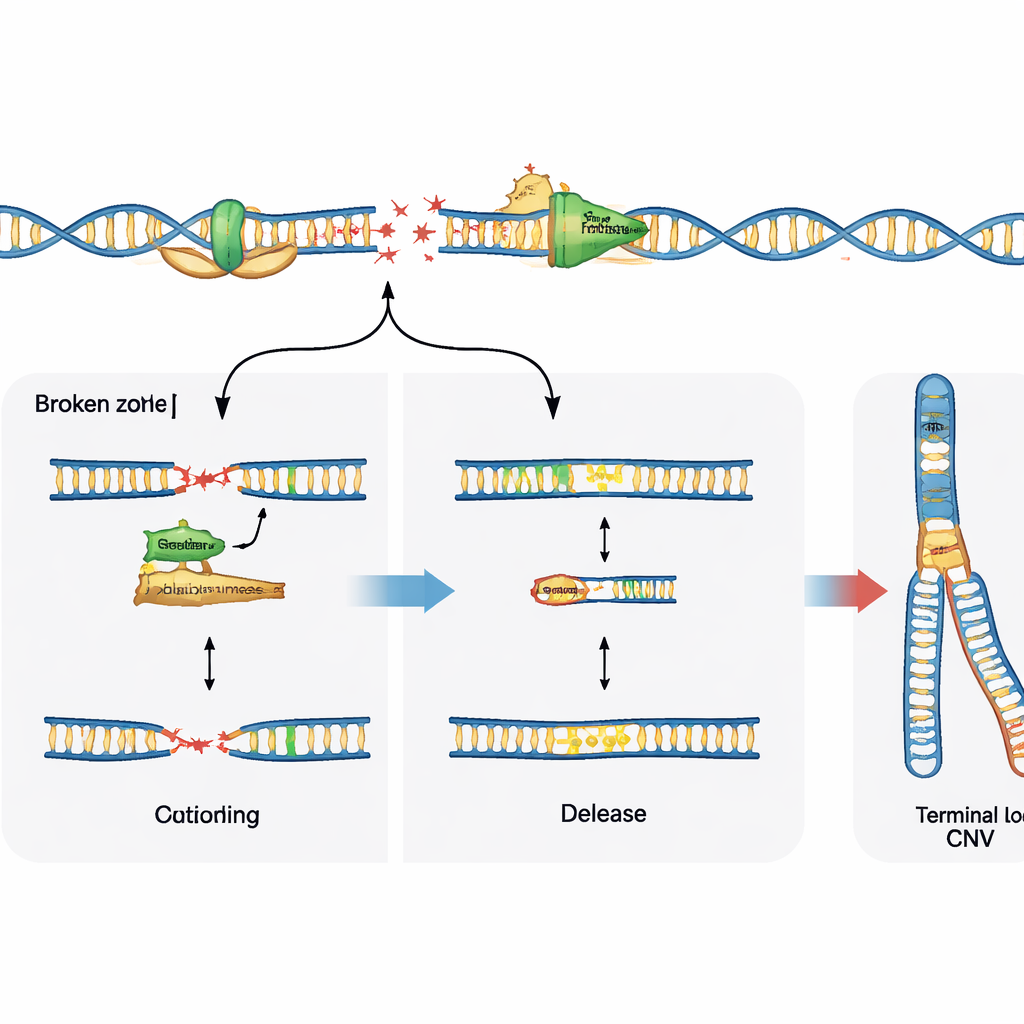
Repair choices steer the outcome
Breaking DNA is only half the story; how the cell repairs those breaks shapes the final pattern of variation. The study probed the role of a specialized enzyme, polymerase theta, which is known to patch and join broken DNA ends. In neural progenitor cells lacking a major repair route called non‑homologous end joining, blocking polymerase theta sharply reduced the usual recurrent deletions at fragile sites, showing that in this setting the enzyme actively promotes copy number changes. In contrast, in normal cells with intact repair systems, inhibiting polymerase theta did not prevent these deletions but instead revealed more raw DNA breaks at fragile sites. This suggests that polymerase theta can either drive genome reshaping when other repair options are missing, or help stabilize forks and limit visible damage when the main repair machinery is present.
Why this matters for brain health and cancer
Taken together, the work paints a clear picture: in neural progenitor cells under replication stress, clustered breaks in long, actively used genes act as central hubs from which both common and rare copy number variants emerge. These structural changes are not fleeting; they become fixed in daughter cells, helping to diversify the genomes of cells in the developing brain and, under the wrong conditions, to feed cancer evolution. By tying together fragile gene regions, replication stress, and specific repair pathways, the study offers a mechanistic bridge between everyday DNA copying errors and the complex patterns of structural variation seen in neurological disease and tumors.
Citation: Corazzi, L., Ing, A., Benito, E. et al. Recurrent DNA break clusters drive replication-stress-induced copy number variants and genome diversification. Nat Commun 17, 3627 (2026). https://doi.org/10.1038/s41467-026-71790-5
Keywords: copy number variants, replication stress, neural progenitor cells, DNA break clusters, genome instability