Clear Sky Science · en
CryoEM structure of mGlu6 captures receptor activation prior to G protein coupling
How Our Eyes Keep Working in the Dark
Seeing in dim light depends on a tiny molecular machine in the retina that never sleeps. This machine, a receptor called mGlu6, sits at the junction between rod photoreceptors and the nerve cells that relay signals to the brain. When mGlu6 fails, people are born with a condition known as congenital stationary night blindness—they cannot see properly in low light, yet their eyes otherwise look normal. This study reveals the three‑dimensional structure of mGlu6 at near‑atomic detail and explains how its motion enables night vision and how inherited mutations disrupt that process.
A Busy Gatekeeper at the First Synapse of Vision
In darkness, rod cells in the retina constantly release the chemical messenger glutamate. This signal is picked up by mGlu6 on the surface of ON‑bipolar cells, which act as the first relay for rod‑driven vision. Active mGlu6 turns on an internal switch—a G protein called Gαo—that keeps an ion channel (TRPM1) closed so the bipolar cell remains quiet. When light hits rods, glutamate release suddenly drops, mGlu6 signaling turns off, the channel opens, and the bipolar cell becomes active, announcing the presence of light. Because this toggle must operate quickly and continuously over a lifetime, the structure of mGlu6 needs to be both stable and exquisitely tuned.
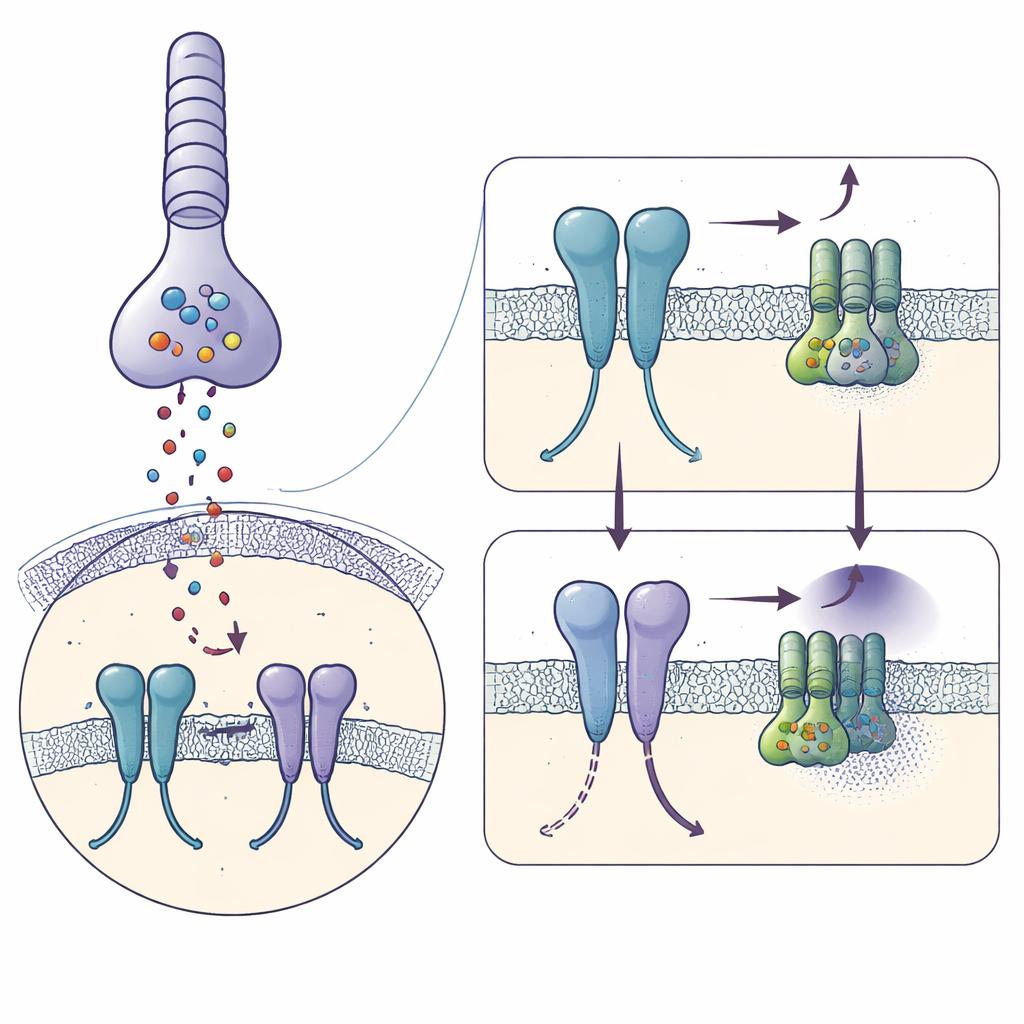
Capturing the Receptor Just Before It Signals
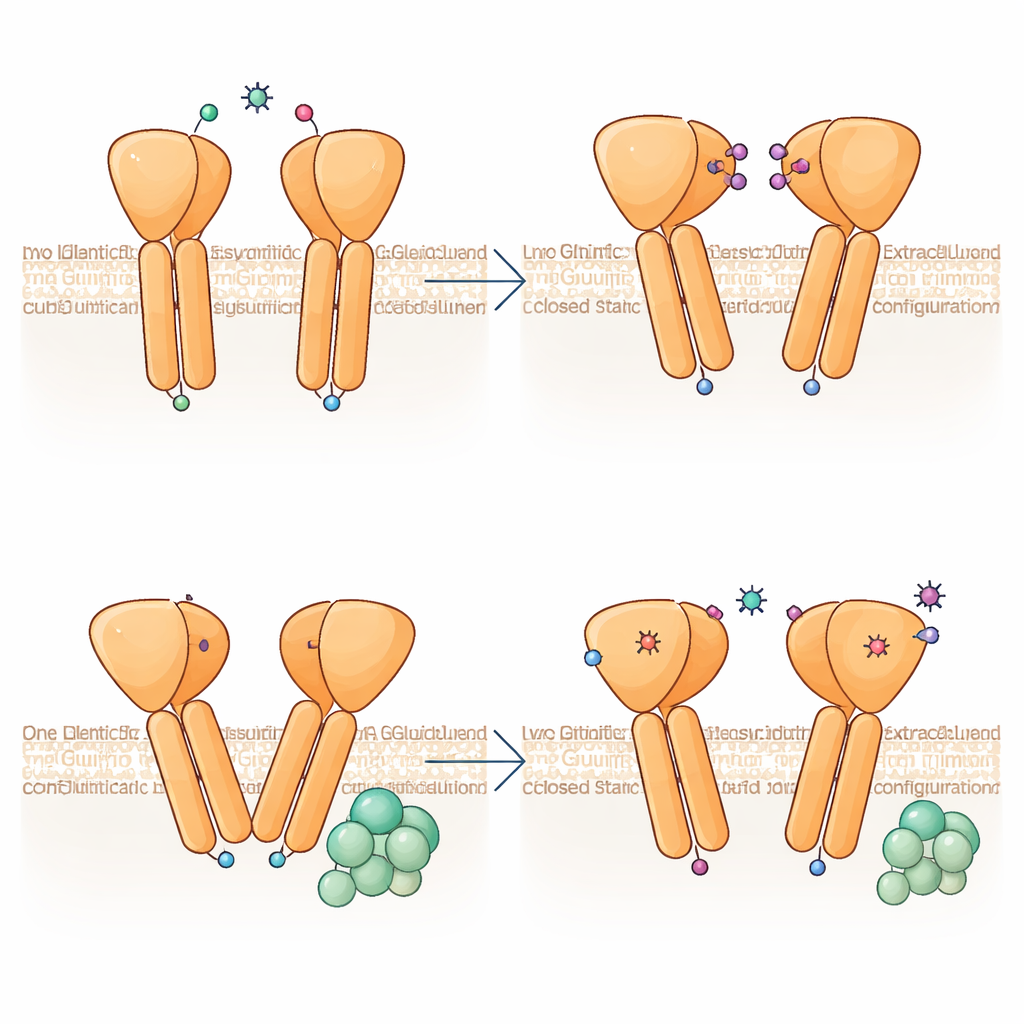
Using cryo‑electron microscopy, the authors froze purified human mGlu6 bound to a strong activator, L‑serine O‑phosphate, and reconstructed its shape at 3.2‑ångström resolution. Each mGlu6 molecule forms a pair, or dimer, made of three stacked regions: a large "clamshell" outside the cell that catches glutamate, a flexible middle segment rich in disulfide bonds, and a seven‑helix bundle spanning the membrane where G proteins bind. The activator clamps the outer domains in a closed, active‑like pose and brings the middle segments close together, while the membrane helices shift so that a key pair of helices (called TM6) come into close contact—hallmarks of a receptor poised to engage a G protein.
Built‑In Asymmetry: One Partner Leads, the Other Follows
A striking discovery is that the two halves of the mGlu6 dimer do not adopt identical shapes, even though both bind the same activator. One subunit bends more sharply, and its membrane helices rise slightly higher toward the outside of the cell, producing an asymmetrical interface where the two TM6 helices meet. This configuration closely resembles the G‑protein‑engaged state seen in related receptors, suggesting that mGlu6 is "pre‑primed" for coupling on one side. Analyses of many particle images show that the dimer samples a continuum of bend angles, from nearly symmetric to strongly asymmetric, implying that the receptor dynamically rocks between states, with asymmetry emerging already before a G protein arrives.
A Hidden Support Scaffold That Speeds Signaling
The work also uncovers an unusual contact surface between the middle cysteine‑rich region and a loop on the outer side of the membrane. In mGlu6, this loop is longer than in related receptors and forms a web of salt bridges, hydrogen bonds, and hydrophobic contacts that lock the middle and membrane regions together. When key residues in this interface are mutated, the receptor still reaches the cell surface and can activate G proteins, but it does so more slowly, even when plenty of receptors are present. This suggests that the extra contacts act as a mechanical brace that efficiently transmits the glutamate‑induced motion of the outer "clamshell" down to the membrane helices, enabling rapid responses needed for real‑time vision.
Explaining Night Blindness Through Structure
By mapping every known patient mutation for congenital stationary night blindness onto the new structure, the authors show how diverse changes can cripple mGlu6. Some mutations distort the glutamate‑binding pocket or disrupt buried hydrophobic cores, destabilizing the outer domain; others break crucial disulfide bonds or salt bridges that hold the middle and membrane regions together, causing the receptor to misfold and fail to reach the cell surface. Many variants reduce the efficiency or speed of G‑protein activation. Intriguingly, two mutations actually boost signaling capacity once expression is normalized, yet still likely cause disease because the receptors are poorly trafficked or mispositioned at synapses. Together, these findings tie specific structural defects to impaired signaling, offering a molecular blueprint for understanding and eventually treating inherited night‑vision disorders.
Citation: Lee, S.Y., Chang, CT., Yun, Y. et al. CryoEM structure of mGlu6 captures receptor activation prior to G protein coupling. Nat Commun 17, 3681 (2026). https://doi.org/10.1038/s41467-026-70436-w
Keywords: night blindness, retinal signaling, G protein–coupled receptors, metabotropic glutamate receptor, cryo electron microscopy