Clear Sky Science · de
Cryo‑EM‑Struktur von mGlu6 zeigt Rezeptoraktivierung vor der G‑Protein‑Kopplung
Wie unsere Augen im Dunkeln weiterarbeiten
Sehen bei schwachem Licht beruht auf einer winzigen molekularen Maschine in der Netzhaut, die niemals ruht. Diese Maschine, ein Rezeptor namens mGlu6, sitzt an der Verbindung zwischen Stäbchenphotorezeptoren und den Nervenzellen, die Signale an das Gehirn weiterleiten. Versagt mGlu6, sind Menschen mit einer als kongenitale stationäre Nachtblindheit bekannten Erkrankung geboren – sie können bei gedämpftem Licht nicht richtig sehen, obwohl ihre Augen sonst normal erscheinen. Diese Studie zeigt die dreidimensionale Struktur von mGlu6 in nahezu atomarer Auflösung und erklärt, wie seine Bewegungen die Nachtsicht ermöglichen und wie vererbte Mutationen diesen Prozess stören.
Ein vielbeschäftigter Wächter an der ersten Synapse des Sehens
Im Dunkeln setzen Stäbchenzellen in der Netzhaut fortlaufend den chemischen Boten Glutamat frei. Dieses Signal wird von mGlu6 auf der Oberfläche der ON‑Bipolarzellen empfangen, die als erste Relaisstation für Stäbchensignale fungieren. Aktiviertes mGlu6 schaltet einen internen Schalter ein – ein G‑Protein namens Gαo –, das einen Ionenkanal (TRPM1) geschlossen hält, sodass die Bipolarzelle ruhig bleibt. Trifft Licht auf die Stäbchen, fällt die Glutamatausschüttung abrupt ab, die mGlu6‑Signalgebung schaltet aus, der Kanal öffnet sich und die Bipolarzelle wird aktiv und meldet das Vorhandensein von Licht. Weil dieser Schalter schnell und über die Lebenszeit hinweg kontinuierlich arbeiten muss, muss die Struktur von mGlu6 sowohl stabil als auch hochpräzise abgestimmt sein.
Den Rezeptor kurz bevor er signalisiert einfangen
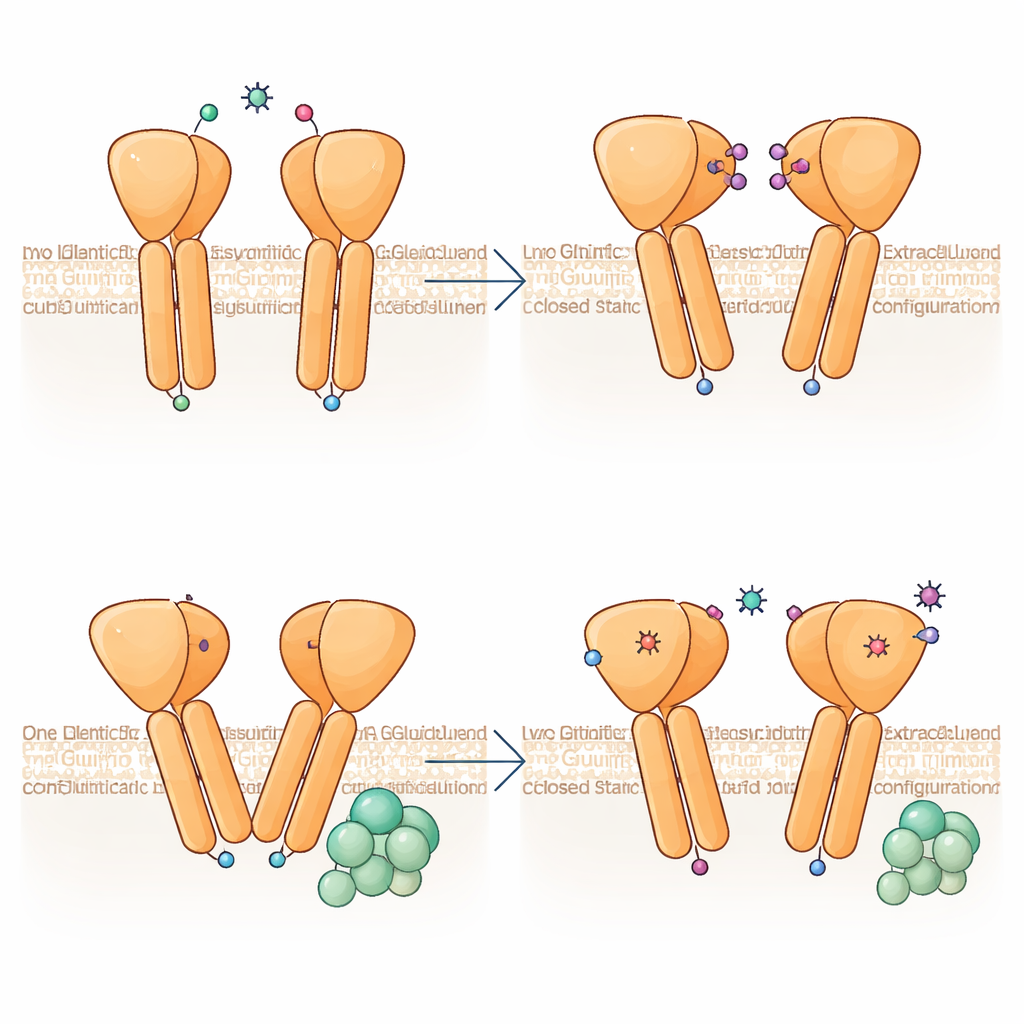
Mithilfe der Kryo‑Elektronenmikroskopie frierten die Autoren gereinigtes menschliches mGlu6 ein, das an einen starken Aktivator, O‑Phosphat‑L‑Serin, gebunden war, und rekonstruierten seine Gestalt mit 3,2 Å Auflösung. Jede mGlu6‑Molekül bildet ein Paar, ein Dimer, bestehend aus drei aufeinandergestapelten Regionen: einer großen äußeren „Muschelschale“, die Glutamat fängt, einem flexiblen mittleren Segment, das reich an Disulfidbrücken ist, und einem siebenhelikalen Bündel, das die Membran durchspannt und an dem G‑Proteine binden. Der Aktivator klemmt die äußeren Domänen in eine geschlossene, aktivitätsähnliche Pose und bringt die mittleren Segmente nahe zusammen, während sich die Membranhelixen so verschieben, dass ein zentrales Helixpaar (genannt TM6) in engen Kontakt kommt – Kennzeichen eines Rezeptors, der bereit ist, ein G‑Protein zu engagieren.
Ein eingebauter Asymmetrie‑Mechanismus: Ein Partner führt, der andere folgt
Eine auffällige Entdeckung ist, dass die beiden Hälften des mGlu6‑Dimers nicht identische Formen annehmen, obwohl beide denselben Aktivator binden. Eine Untereinheit knickt deutlich schärfer, und ihre Membranhelixen ragen etwas weiter nach außen, wodurch eine asymmetrische Schnittstelle entsteht, an der die beiden TM6‑Helices zusammentreffen. Diese Konfiguration ähnelt stark dem G‑Protein‑gebundenen Zustand, der bei verwandten Rezeptoren beobachtet wurde, und legt nahe, dass mGlu6 auf einer Seite bereits „vor‑eingestellt“ für die Kopplung ist. Analysen zahlreicher Partikelbilder zeigen, dass das Dimer ein Kontinuum von Knickwinkeln durchmisst, von nahezu symmetrisch bis stark asymmetrisch, was darauf hindeutet, dass der Rezeptor dynamisch zwischen Zuständen schaukelt und die Asymmetrie bereits vor Ankunft eines G‑Proteins entsteht.
Ein verborgenes Stützgerüst, das die Signalgebung beschleunigt
Die Arbeit deckt außerdem eine ungewöhnliche Kontaktfläche zwischen der mittleren cysteinreichen Region und einer Schleife auf der Außenseite der Membran auf. Bei mGlu6 ist diese Schleife länger als bei verwandten Rezeptoren und bildet ein Netz aus Salzbrücken, Wasserstoffbrücken und hydrophoben Kontakten, das die mittleren und membranständigen Regionen zusammen verriegelt. Werden Schlüsselfunktionen in dieser Schnittstelle mutiert, erreicht der Rezeptor zwar noch die Zelloberfläche und kann G‑Proteine aktivieren, tut dies aber langsamer, selbst wenn viele Rezeptoren vorhanden sind. Das deutet darauf hin, dass die zusätzlichen Kontakte als mechanische Verstrebung wirken, die die durch Glutamat ausgelöste Bewegung der äußeren „Muschelschale“ effizient auf die Membranhelices überträgt und so die schnellen Reaktionen ermöglicht, die für Echtzeit‑Sehen nötig sind.
Nachtblindheit durch Struktur erklärt
Indem die Autoren jede bekannte Patientenmutation bei kongenitaler stationärer Nachtblindheit auf die neue Struktur abbilden, zeigen sie, wie vielfältige Veränderungen mGlu6 beeinträchtigen können. Manche Mutationen verzerren die Glutamatbindetasche oder stören eingekapselte hydrophobe Kerne und destabilisieren die äußere Domäne; andere zerbrechen wichtige Disulfidbrücken oder Salzbrücken, die die mittleren und membranständigen Regionen zusammenhalten, sodass der Rezeptor falsch faltet und nicht zur Zelloberfläche gelangt. Viele Varianten verringern die Effizienz oder die Geschwindigkeit der G‑Protein‑Aktivierung. Intriguierenderweise steigern zwei Mutationen bei normalisierter Expression tatsächlich die Signalstärke, verursachen aber vermutlich dennoch Krankheit, weil die Rezeptoren schlecht transportiert werden oder an Synapsen fehlpositioniert sind. Insgesamt verknüpfen diese Befunde spezifische strukturelle Defekte mit gestörter Signalübertragung und liefern einen molekularen Bauplan, um vererbte Sehstörungen bei Dunkelheit besser zu verstehen und schließlich zu behandeln.
Zitation: Lee, S.Y., Chang, CT., Yun, Y. et al. CryoEM structure of mGlu6 captures receptor activation prior to G protein coupling. Nat Commun 17, 3681 (2026). https://doi.org/10.1038/s41467-026-70436-w
Schlüsselwörter: Nachtblindheit, retinale Signalübertragung, G‑Protein‑gekoppelte Rezeptoren, metabotroper Glutamatrezeptor, Kryo‑Elektronenmikroskopie