Clear Sky Science · en
ATRX loss couples genome instability at a G-rich repeat to dysregulation of human alpha-globin expression
Why this matters for blood and cancer
Our red blood cells depend on finely tuned control of the genes that make haemoglobin, the protein that carries oxygen. A protein called ATRX sits on DNA and helps keep it stable, and mutations in ATRX cause a form of inherited anaemia with intellectual disability and are common in several cancers. This study uses the alpha-globin gene cluster as a test case to show, in molecular detail, how losing ATRX can damage DNA at a repetitive stretch and quietly switch off nearby genes. Understanding this chain of events helps explain a rare human disorder and sheds light on how genome instability can alter gene activity more broadly.
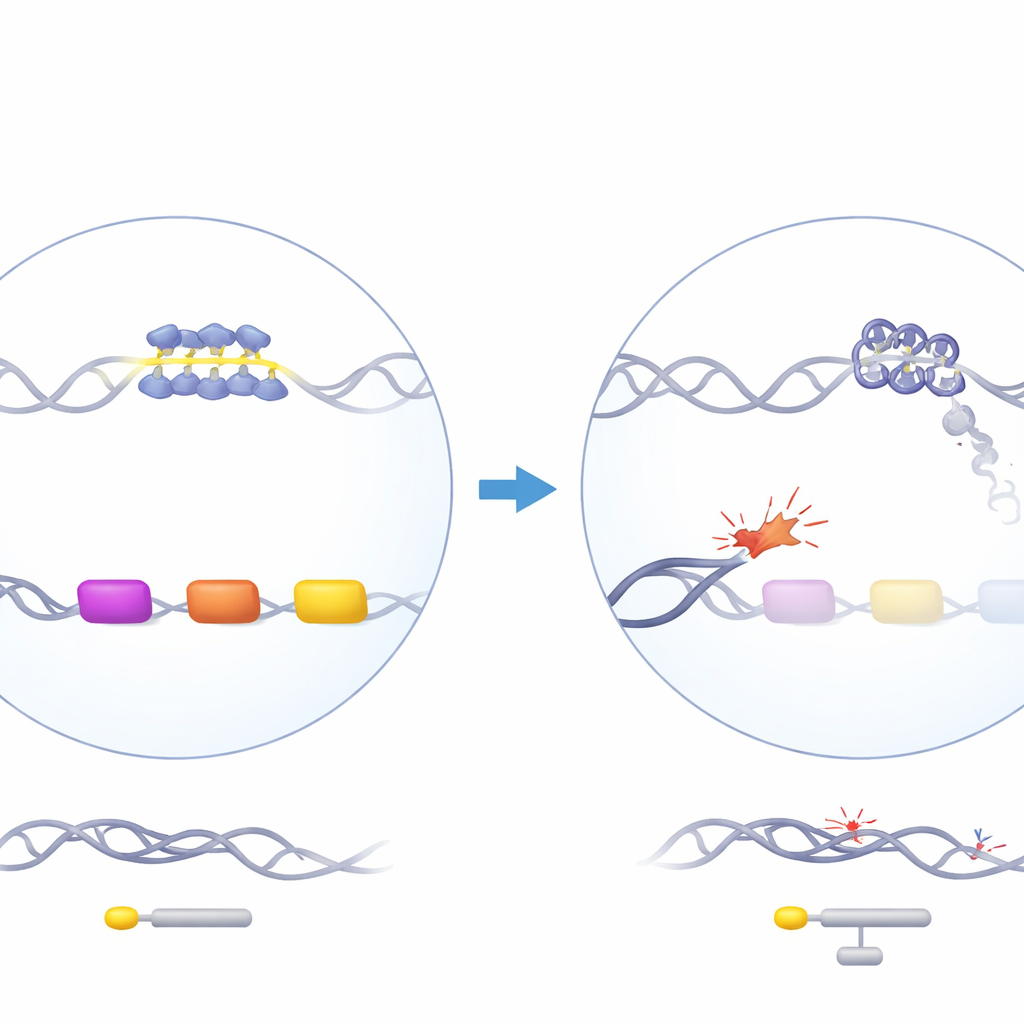
A guardian protein with a puzzling effect
ATRX is a chromatin remodelling protein that helps organise DNA and is involved in replication, transcription and DNA repair. When ATRX is mutated in humans, patients develop ATR-X syndrome, which includes mild alpha-thalassemia: their alpha-globin genes, needed to build haemoglobin, are underactive. Intriguingly, ATRX loss affects alpha-globin but not beta-globin, even though both are required in equal amounts. The alpha-globin genes sit in a gene-rich, GC-rich region of chromosome 16 that contains several repetitive DNA elements, including a particularly G-rich variable number tandem repeat (VNTR) located just upstream of the alpha-like globin genes. Earlier genetic work showed that the length of this repeat correlates with how strongly alpha-globin is reduced in patients, hinting that this odd stretch of DNA might be central to the problem.
Zooming in on the affected cells
To investigate, the authors removed ATRX in human blood stem and progenitor cells using CRISPR gene editing and then matured these cells into red blood cell precursors in culture. Looking at the cell population as a whole showed only a mild drop in one alpha-like gene, HBM, and no major change in other globin genes, mirroring the modest blood changes seen in patients. However, when they analysed single colonies and single cells, a different picture emerged. Only a subset of ATRX-deficient cells showed strong reduction of HBM and, to a lesser degree, the main alpha-globin genes. These same cells displayed signatures of an activated DNA damage response, including increased markers of broken DNA and heightened modification of a histone protein (H2A) that is linked to repairing damage and silencing nearby genes. This suggested that alpha-globin downregulation occurs mainly in cells where ATRX loss has allowed local DNA damage to accumulate.
A troublesome G-rich repeat as the weak spot
The team then focused on the G-rich VNTR embedded in a nearby pseudogene called HBZP1. Using an immortalised human erythroid cell line, they tracked ATRX binding across the alpha-globin region and found that ATRX is specifically recruited to this VNTR when the surrounding DNA is being transcribed. They engineered a system where ATRX could be rapidly degraded and observed that losing ATRX reduced HBM expression, again mimicking the disease phenotype. Strikingly, when they deleted the VNTR itself from the genome, removing ATRX no longer lowered HBM levels. Drugs that stabilise unusual four-stranded DNA structures known as G-quadruplexes produced a similar drop in HBM expression, but only when the VNTR was present. Genome-wide analyses revealed that many other genes misregulated by ATRX loss also contain GC-rich repetitive elements, pointing to a general vulnerability at such sites.
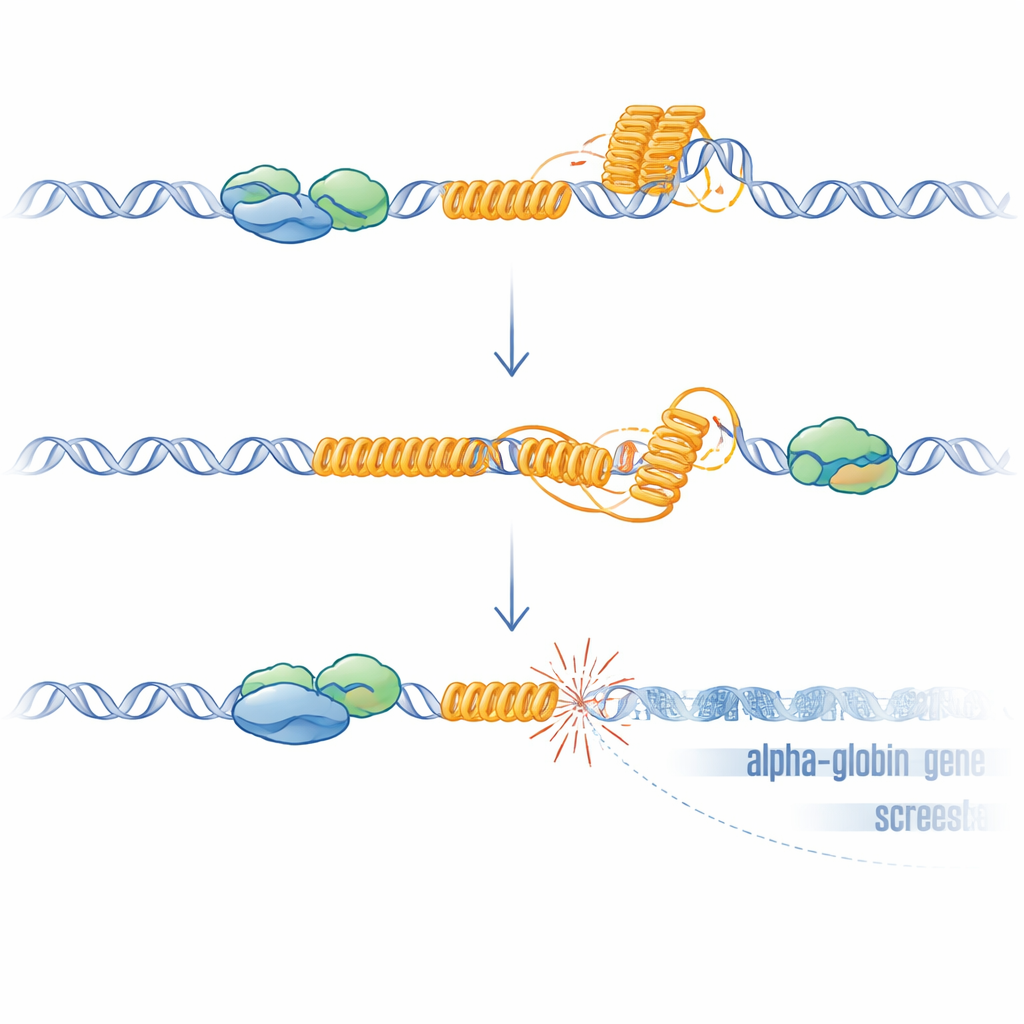
From odd DNA shapes to broken genes
G-rich repeats like this VNTR can fold into G-quadruplexes and foster the formation of R-loops, hybrid structures where newly made RNA remains stuck to the DNA template. Both structures can slow or stall the machinery that copies and reads DNA, causing stress and breaks. The researchers showed that, without ATRX, R-loops increased across the genome and were particularly elevated at the VNTR. When they overexpressed RNase H, an enzyme that removes R-loops, HBM expression partially recovered, supporting a causal role for these structures. To test whether DNA damage alone was sufficient to silence the nearby genes, they used CRISPR to create targeted breaks just upstream of HBM in cells where the VNTR had been deleted. These artificial breaks, even in the absence of the repeat, again reduced HBM and, more weakly, the alpha-globin genes, with the closest gene being most affected. This distance-dependent effect matches what is seen in patients and is consistent with known spreading of transcriptional repression away from sites of double-strand breaks.
What this means for disease and beyond
Taken together, the work outlines a clear model: in healthy cells, ATRX binds to a G-rich repeat near the alpha-globin genes as it is transcribed, helping to prevent or dissolve G-quadruplexes and R-loops and thereby protecting the region from DNA damage. When ATRX is missing, these unusual DNA and RNA structures accumulate at the repeat, leading to local DNA breaks, activation of a damage response, and a repressive chromatin environment that dampens expression of nearby genes in a distance-dependent fashion, with HBM hit hardest. This mechanism explains why ATR-X syndrome causes only mild, patchy alpha-thalassemia, why the severity varies with repeat length, and why mouse models lacking this human-specific repeat do not show the same blood defect. More broadly, it suggests that repetitive, G-rich sequences scattered across our genome can act as hidden fault lines: if protective factors like ATRX fail, they can become hotspots of instability that quietly rewire gene activity in development, ageing and cancer.
Citation: Shen, Y., Gupta, K., Tan-Wong, S.M. et al. ATRX loss couples genome instability at a G-rich repeat to dysregulation of human alpha-globin expression. Nat Commun 17, 2749 (2026). https://doi.org/10.1038/s41467-026-69169-7
Keywords: ATRX, alpha-globin, DNA damage, G-quadruplex, R-loops