Clear Sky Science · en
cGAS-STING/HMGB1-mediated senescence induced by LRRK2 accelerates cartilage degeneration in osteoarthritis
Why worn-out joints matter
Achy knees and stiff hips are often written off as an unavoidable part of getting older, but osteoarthritis is more than simple "wear and tear." It involves living cells inside cartilage that gradually change, stop repairing tissue, and start sending out distress signals that inflame the joint. This study uncovers a new molecular culprit that helps push these cells into a prematurely aged state, offering a fresh angle for understanding—and potentially slowing—the progression of osteoarthritis.
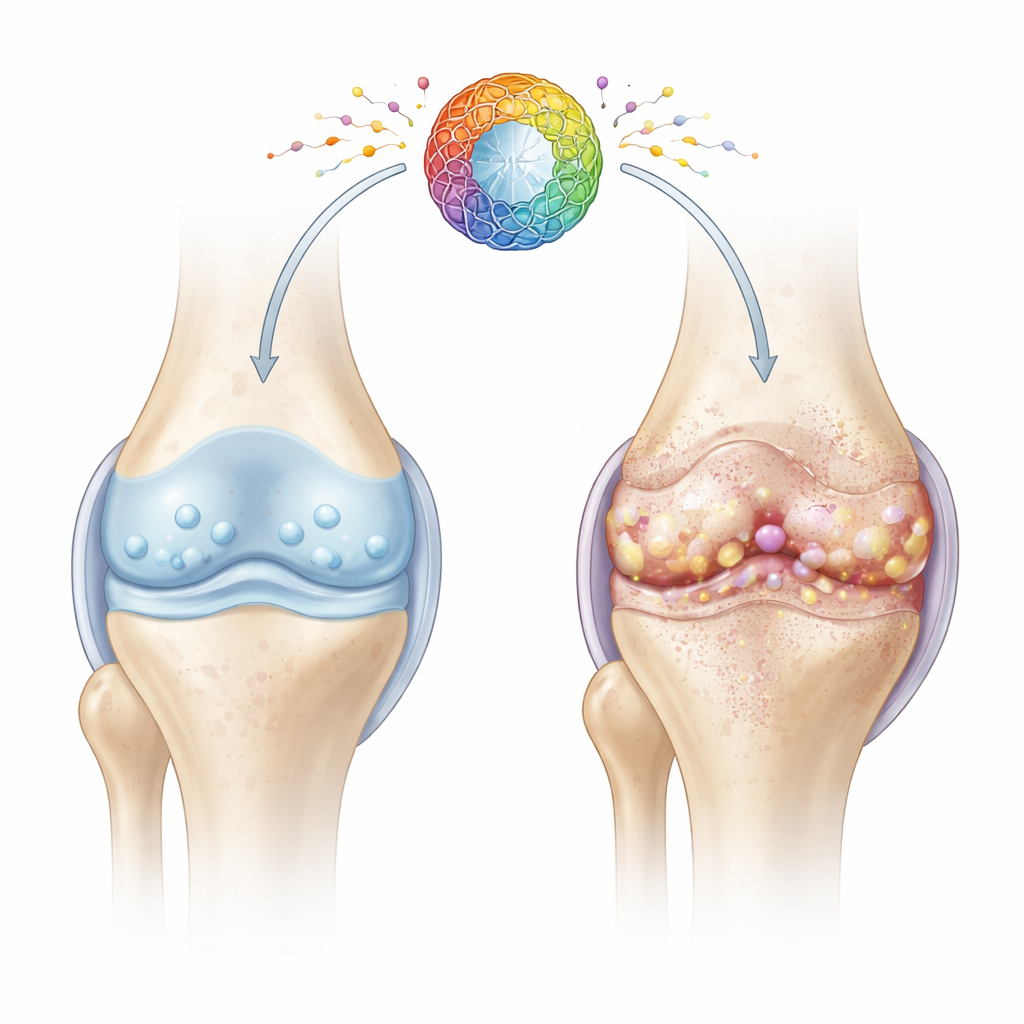
A closer look at tired joint cells
Cartilage is the smooth, rubbery tissue that cushions the ends of bones. Its health depends on chondrocytes, specialized cells that quietly maintain the cartilage matrix for decades. With age and injury, many chondrocytes enter a state called cellular senescence: they stop dividing, lose their repair capacity, and release a cocktail of inflammatory and tissue-damaging molecules. The authors focused on how problems in the cells’ energy factories—mitochondria—might drive this harmful aging state and speed up joint degeneration.
A Parkinson’s-linked enzyme shows up in sore joints
Using public gene expression data from human cartilage, the team searched for genes tied to both osteoarthritis and cellular aging. One stood out: LRRK2, a large enzyme better known for its role in inherited Parkinson’s disease and inflammatory disorders. LRRK2 levels were markedly higher in osteoarthritic cartilage compared with healthy tissue. In a rat model of knee osteoarthritis, LRRK2 was similarly elevated in joint cartilage and lining cells. When the researchers artificially boosted LRRK2 in rat chondrocytes, hundreds of other genes shifted, especially those involved in cell aging and inflammatory signaling, suggesting that LRRK2 is not just a bystander but an active driver of the disease process.
How internal alarms turn stress into damage
The study then zoomed in on a molecular alarm system inside cells called the cGAS–STING pathway, which detects misplaced DNA in the cell fluid—a frequent consequence of mitochondrial damage—and triggers inflammation. Laboratory experiments showed that excess LRRK2 binds to and activates this pathway in chondrocytes. Activation, in turn, raised the levels and altered the behavior of HMGB1, a protein that usually helps organize DNA in the nucleus but can act as a powerful danger signal when released into the cell’s surroundings. Chondrocytes with high LRRK2 showed swollen, malfunctioning mitochondria, more reactive oxygen species (damaging oxygen byproducts), reduced energy production, and clear hallmarks of senescence. Blocking STING, or inhibiting LRRK2’s activity or its GTPase function, dampened HMGB1, eased oxidative stress, and reduced aging markers in these cells.
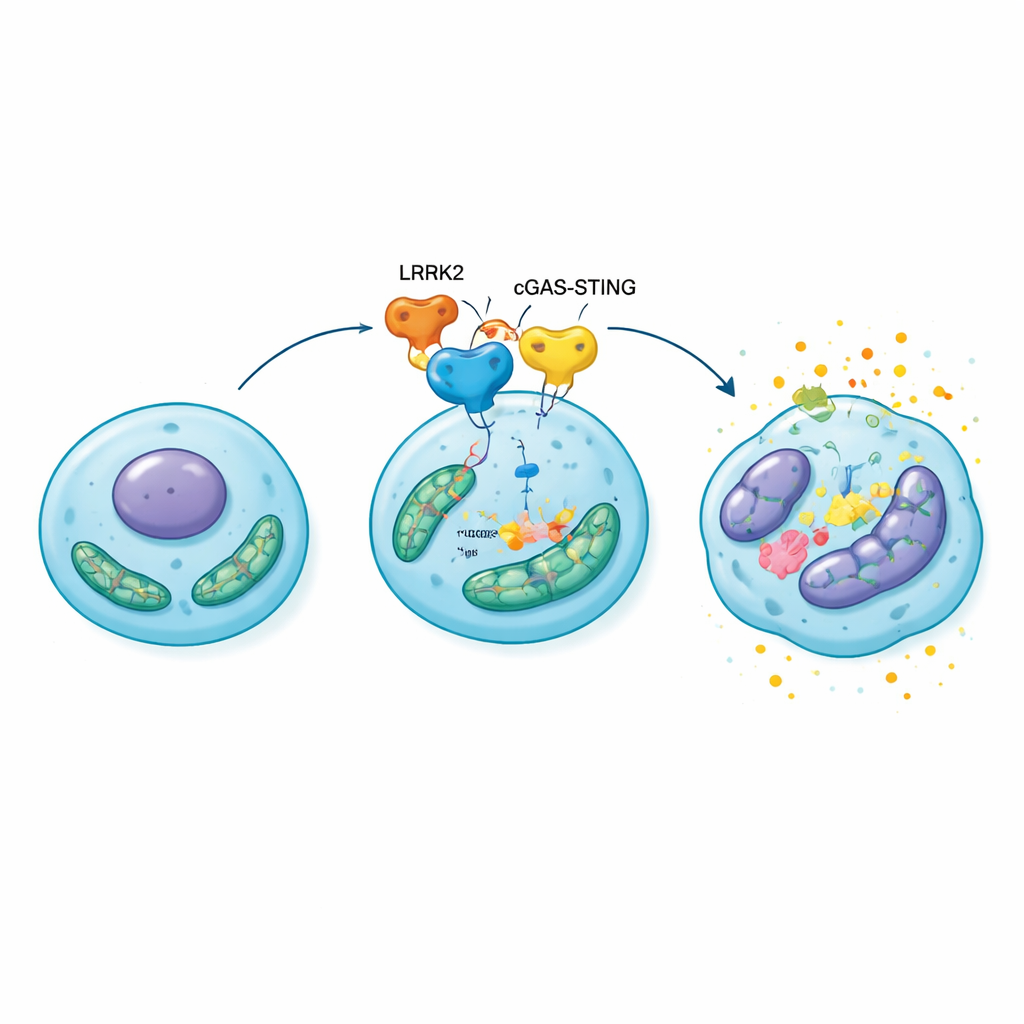
From stressed cells to crumbling cartilage
To see whether this molecular chain reaction mattered for whole joints, the researchers overexpressed LRRK2 directly in the knees of rats with surgically induced osteoarthritis. Compared with control animals, these rats developed thinner, more eroded cartilage, greater bone changes beneath the joint surface, and more senescent chondrocytes loaded with inflammatory and matrix-degrading molecules. When the same rats were treated with a STING-blocking drug, joint damage and cell aging were noticeably reduced, and mitochondrial function partly recovered. These results place the LRRK2–cGAS–STING–HMGB1 route at the center of a self-reinforcing loop: mitochondrial stress activates the alarm system, which boosts HMGB1 and inflammation, which in turn deepens mitochondrial injury and pushes more chondrocytes into senescence.
What this means for future joint care
In plain terms, the authors show that LRRK2 acts like a faulty switch that locks cartilage cells into an old, exhausted state and accelerates the breakdown of joint tissue. By flipping on the cGAS–STING–HMGB1 alarm network, high LRRK2 activity links internal cellular stress to chronic inflammation and structural damage in osteoarthritis. This work suggests that drugs aimed at dialing down LRRK2 or interrupting the cGAS–STING pathway could help keep chondrocytes functional for longer, potentially slowing the progression of osteoarthritis rather than just easing its symptoms.
Citation: Zhang, Y., Zhu, Z., Ji, P. et al. cGAS-STING/HMGB1-mediated senescence induced by LRRK2 accelerates cartilage degeneration in osteoarthritis. Cell Death Dis 17, 377 (2026). https://doi.org/10.1038/s41419-026-08651-y
Keywords: osteochondral degeneration, cellular senescence, mitochondrial dysfunction, inflammatory signaling, LRRK2 pathway