Clear Sky Science · en
Ubiquitin-specific protease 26 facilitates endochondral ossification by driving chondrocyte hypertrophy and mineralization
How Bones Grow and Repair Themselves
Our skeleton is constantly changing. Long bones in the arms and legs first form as soft cartilage, which later turns into hard bone in a process called endochondral ossification. The same program helps broken bones heal—but when it runs too strongly in joints, it can also fuel osteoarthritis. This study uncovers a key molecular switch, a protein called USP26, that helps cartilage cells grow, harden, and respond to mechanical forces such as body weight and movement. Understanding this switch could open new ways to boost bone repair while slowing joint damage.
The Journey from Soft Cartilage to Hard Bone
In growing embryos and healing fractures, specialized cartilage cells known as chondrocytes first multiply, then enlarge, and finally help lay down mineral to form bone. The authors found that USP26 becomes more abundant exactly when and where this transition is happening: in the middle of developing mouse limb bones, in the cartilage callus that bridges a healing fracture, and in overgrown cartilage areas in osteoarthritic joints. When chondrocytes are encouraged to mature in the lab, USP26 levels rise in step with classic markers of hypertrophy (cell enlargement) and mineralization. These patterns suggest that USP26 acts as an accelerator for the cartilage-to-bone transition. 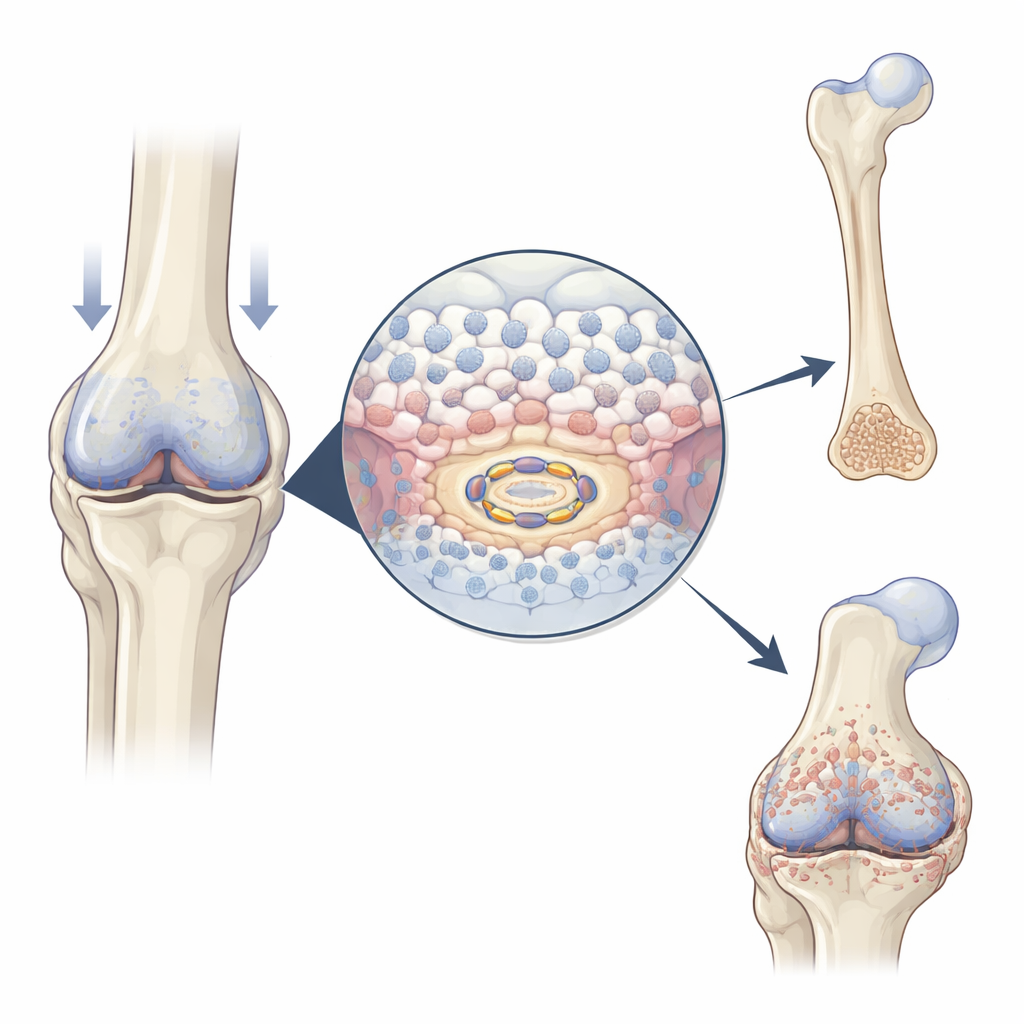
What Happens When USP26 Is Missing
To test USP26’s role, the team engineered mice in which this protein was removed only from chondrocytes. These animals developed shorter limbs and vertebrae, reflecting stunted skeletal growth. Their growth plates—the zones where new bone is normally made—showed fewer enlarged chondrocytes, less mineralized cartilage, and reduced expression of key genes that drive bone formation and blood vessel invasion. When these knockout mice suffered femur fractures, their calluses contained less cartilage and bone, and scans revealed weaker, less mineralized repair tissue. At the same time, removing USP26 from joint cartilage protected against experimentally induced osteoarthritis: there were fewer bony spurs, smoother cartilage surfaces, and lower scores of disease severity. Together, the findings show that USP26 is double-edged—it is needed for normal growth and repair, but it also contributes to harmful bone overgrowth in diseased joints.
Fueling Bone Formation Through Cellular Power Plants
Chondrocyte enlargement and mineralization demand large amounts of energy. The researchers discovered that USP26 helps meet this demand by maintaining healthy mitochondria, the cell’s power plants. Without USP26, chondrocytes took up less glucose, made less ATP (the cell’s energy currency), and produced less lactate. Their mitochondria were fewer, more fragmented, and functioned poorly, with reduced respiratory activity and expression of genes involved in oxidative phosphorylation. A protein called FBP2 emerged as a crucial link: in the absence of USP26, FBP2 levels rose sharply. FBP2 is known to tilt cells away from burning glucose and to suppress mitochondrial biogenesis. Here, blocking FBP2 in USP26-deficient chondrocytes restored glucose use, mitochondrial mass, and energy production, and revived their ability to enlarge and mineralize. In osteoarthritis models, inhibiting FBP2 also intensified cartilage ossification, underscoring how the USP26–FBP2 balance shapes bone-related outcomes. 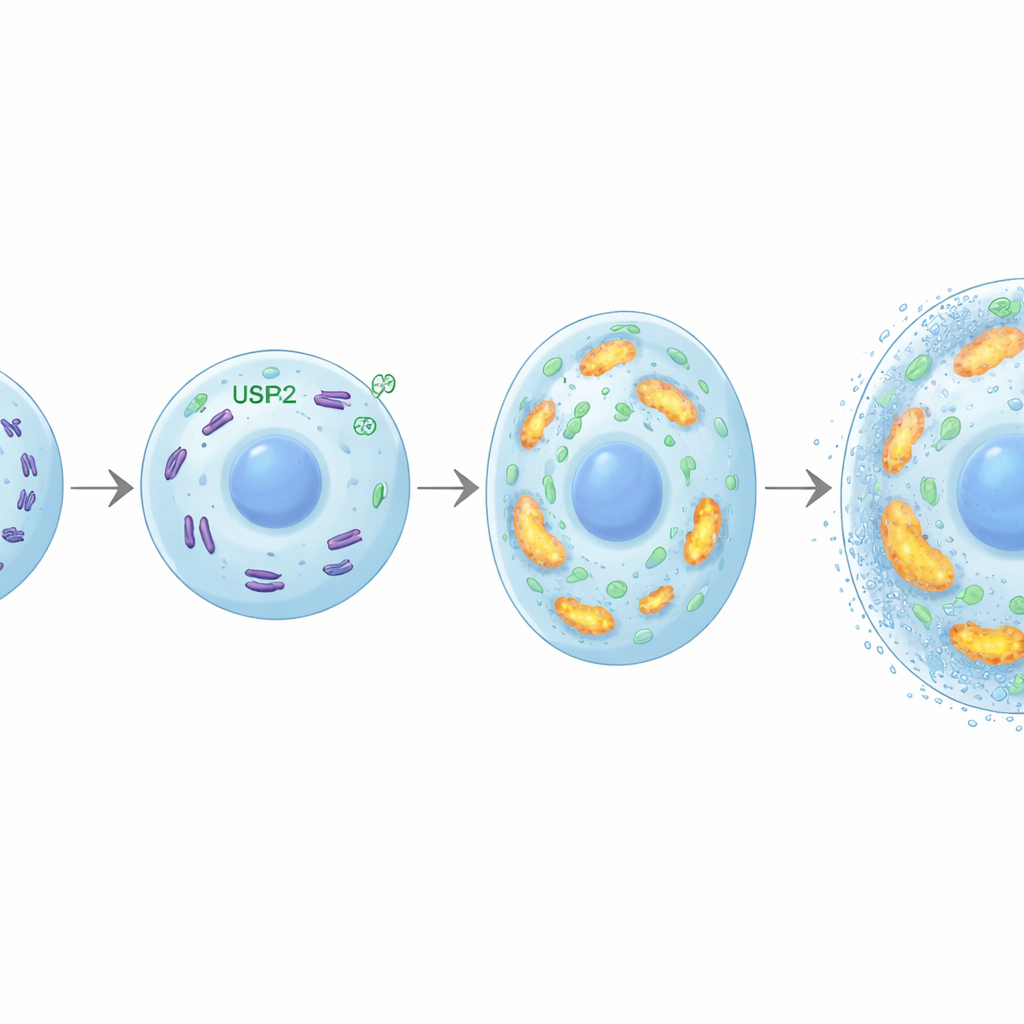
How Mechanical Forces Turn On USP26
The skeleton constantly senses and adapts to mechanical forces. The team found that zones of cartilage under heavy load—such as weight-bearing areas in arthritic knees—show higher USP26 levels. In cultured chondrocytes, applying controlled compression boosted USP26 and simultaneously turned on genes that drive hypertrophy and mineralization. This mechanical signal flowed through estrogen receptor‑α (ER‑α), a hormone-responsive transcription factor. Under compression, ER‑α became activated by phosphorylation at a specific site (serine 118) and bound directly to a short DNA sequence in the USP26 gene’s control region. Mutating either this DNA site or the phosphorylation site on ER‑α blunted the rise in USP26, preserved higher FBP2 levels, and weakened the metabolic and developmental shift toward bone formation. In mice forced to run and overload their knee joints, deletion of USP26 in chondrocytes again reduced bony overgrowth and cartilage damage, confirming its role as a mechanical “sensor–effector” in vivo.
Why This Matters for Bones and Joints
Put simply, USP26 helps cartilage cells convert mechanical load and fuel supplies into bone-building activity. It does this by lowering FBP2, preserving robust, energy-producing mitochondria, and pushing chondrocytes toward enlargement and mineralization. That makes USP26 essential for normal skeletal growth and effective fracture healing. But in aging or injured joints exposed to chronic overload, the same pathway appears to drive the hardening and overgrowth of cartilage that mark osteoarthritis. By targeting USP26 itself, or its downstream partner FBP2, future therapies might one day selectively amplify endochondral ossification when we need more bone—after trauma or in growth disorders—while dialing it down in joints at risk of degenerative disease.
Citation: Li, C., Xu, Y., Zhou, L. et al. Ubiquitin-specific protease 26 facilitates endochondral ossification by driving chondrocyte hypertrophy and mineralization. Bone Res 14, 41 (2026). https://doi.org/10.1038/s41413-026-00517-5
Keywords: endochondral ossification, chondrocyte hypertrophy, mitochondrial metabolism, mechanotransduction, osteoarthritis