Clear Sky Science · en
Osteoblast-derived osteomodulin restrains osteoclastogenesis via ITGB8/RRM2-mediated reduction of mitochondrial respiration and mitochondrial ATP production
Why this matters for bones
Osteoporosis quietly weakens bones in millions of older adults, especially women after menopause. Most current drugs work by broadly slowing bone breakdown or boosting bone formation, often with side effects. This study uncovers a natural "brake" built into bone itself: a protein made by bone-building cells that tells bone-eating cells to dial down their energy use, and thus their destructive power. Understanding this built‑in safety system could inspire safer, more targeted osteoporosis treatments.
A hidden signal between bone cells
Bone health depends on a constant tug-of-war between two main cell types. Osteoblasts build new bone, while osteoclasts dissolve old bone so it can be replaced. The authors focused on an overlooked signal called osteomodulin, or OMD, a small protein secreted by osteoblasts into the bone matrix. By mining several large genetic datasets from different osteoporosis models, they found that the gene for OMD was consistently turned down in fragile bones. When they examined bone tissue and blood from postmenopausal women, those with osteoporosis had less OMD in both bone and circulation, and lower OMD levels went hand in hand with markers of rapid bone loss. Similar reductions appeared in a mouse model of estrogen-loss osteoporosis, suggesting that OMD decline is a common feature of weakening bone. 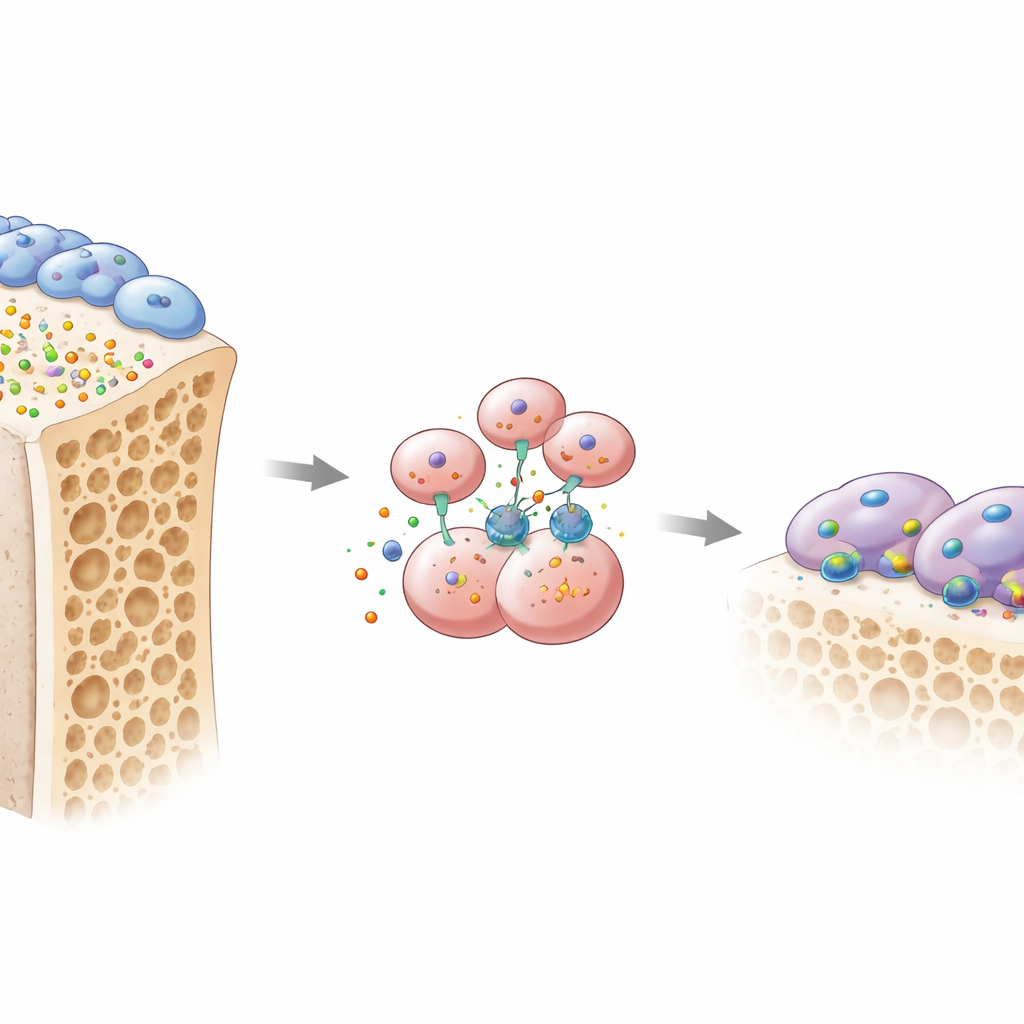
What happens when the brake is removed
To test cause and effect, the team engineered mice lacking OMD. When OMD was deleted throughout the body, or specifically in osteoblasts, the animals lost much more spongy bone and showed a surge in active osteoclasts chewing along bone surfaces. In contrast, removing OMD only from osteoclast precursors themselves had little impact, implying that OMD normally acts as a message sent from osteoblasts to their bone-resorbing neighbors. In lab dishes, osteoclast precursors grown with osteoblasts that made little OMD turned into large, aggressive osteoclasts. Adding back purified OMD protein flipped the script: it sharply reduced formation of these multinuclear bone-eating cells and disrupted their specialized "sealing ring" structures that they use to grip and dissolve bone.
Turning down the cellular power plants
The researchers then asked how this osteoblast signal could so powerfully restrain osteoclasts. Their analyses pointed to energy metabolism. Osteoclasts are energy-hungry and rely heavily on mitochondria, the cell’s power plants, to generate ATP. When osteoclast precursors were exposed to OMD, genes and proteins involved in mitochondrial respiration dropped, mitochondrial DNA copies declined, and direct measurements showed lower oxygen use and ATP production by mitochondria. Glycolysis, a backup sugar-burning pathway, rose only slightly and could not fully compensate. As a result, the cells’ mitochondrial membrane potential fell and their overall metabolic profile shifted toward an energy-stressed state. These changes were closely tied to reduced levels of a key enzyme subunit called RRM2, which helps supply the DNA building blocks required to maintain mitochondrial DNA.
A step‑by‑step chain from surface sensor to genes
Diving deeper, the team traced a signaling chain that links an outside protein to inner energy control. OMD was found to bind a specific receptor, integrin β8, on osteoclast precursors. This interaction dampened the activity of a molecular switch, RhoA, and increased the tagging (phosphorylation) of the regulator YAP, causing YAP to leave the nucleus. Without YAP partnering with TEAD proteins on DNA, the Rrm2 gene quieted down. Lower RRM2 meant fewer DNA building blocks for mitochondria, less mitochondrial DNA, reduced abundance of electron transport chain components, and ultimately weaker mitochondrial ATP output. When the researchers blocked RRM2 with a small molecule, osteoclast formation and mitochondrial function declined much like they did under OMD. Conversely, forcing cells to make extra RRM2 partially rescued their energy production and osteoclast development even in the presence of OMD. 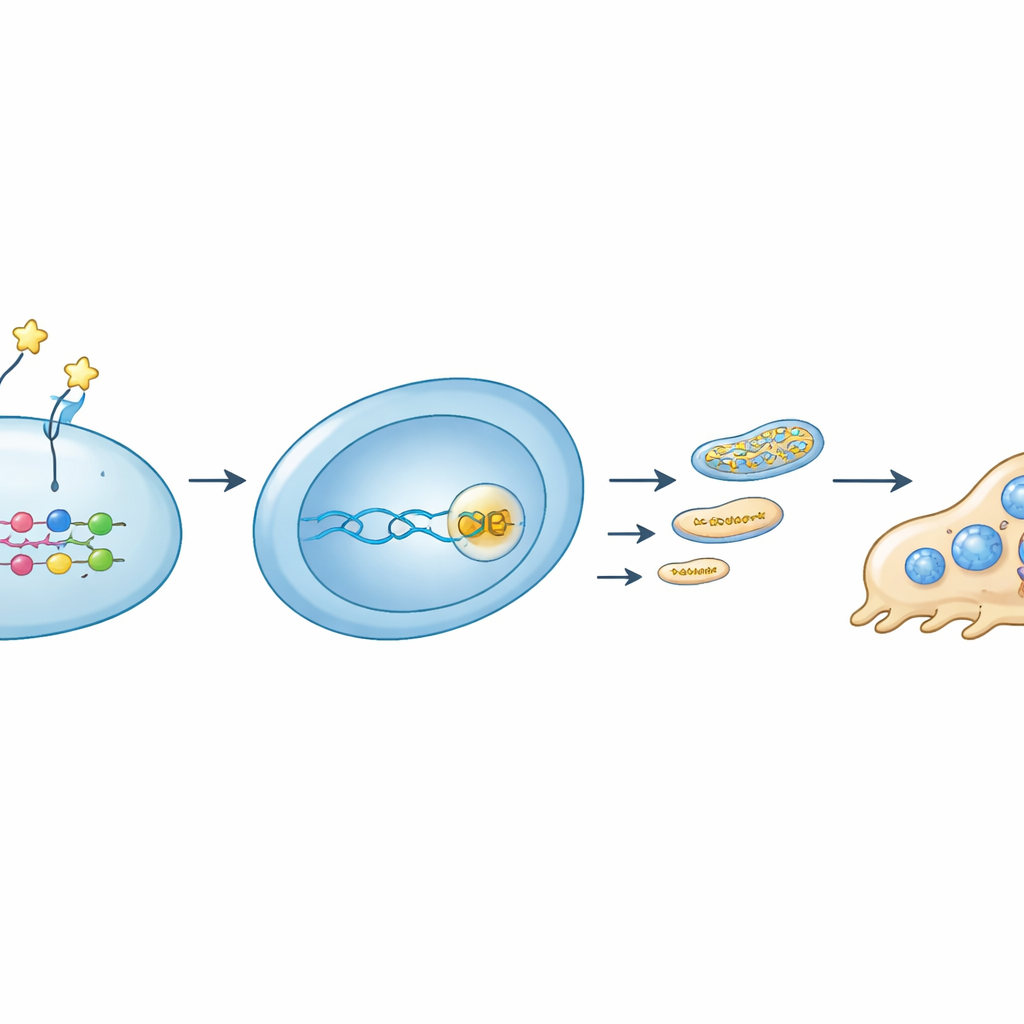
Testing potential new treatment angles
Finally, the study asked whether boosting this natural brake could protect bones in living animals. In mice that had their ovaries removed to mimic postmenopausal bone loss, regular injections of recombinant OMD protein or the RRM2‑blocking drug osalmid both preserved bone structure. Treated mice had denser trabecular bone, fewer osteoclasts, and lower levels of mitochondrial proteins within those osteoclasts. Similar benefits appeared in a model of rapid, inflammation-driven bone loss triggered by bacterial toxins. Importantly, short-term treatment did not visibly harm major organs, although longer and more detailed safety testing will be needed.
What this means for future care
Taken together, the work reveals that osteomodulin acts as a crucial peacekeeper in bone. When produced by osteoblasts, it signals through integrin β8 to reprogram osteoclast precursors, throttling their mitochondrial power supply so they cannot overgrow or overwork. In osteoporosis, this brake appears to weaken, allowing bone-resorbing cells to become more energetic and destructive. By restoring OMD or targeting the downstream energy enzyme RRM2, it may be possible to gently slow bone breakdown without shutting it off completely, offering a new, metabolism-focused strategy to protect aging skeletons.
Citation: Jiang, X., Chen, H., Hou, W. et al. Osteoblast-derived osteomodulin restrains osteoclastogenesis via ITGB8/RRM2-mediated reduction of mitochondrial respiration and mitochondrial ATP production. Exp Mol Med 58, 879–897 (2026). https://doi.org/10.1038/s12276-026-01682-7
Keywords: osteoporosis, bone remodeling, osteoclasts, mitochondrial metabolism, osteomodulin