Clear Sky Science · zh
通过插补和特征选择在高维蛋白质生物标志物数据中提升生存风险预测
这对患者为何重要
医生越来越希望用血液检测来预测个体肿瘤的可能进展——是否会复发或转移——并据此制定个性化治疗方案。现代蛋白检测能够同时测量数百种分子,但产生的数据通常很混乱,包含大量缺失值,并且测量数量远超患者样本数。本文展示了如何小心清理和分析此类复杂数据,使生存预测更可靠且更便于临床人员解读。
把混乱的实验室结果变为有用信号
作者关注蛋白质生物标志物,即血液中某些分子的水平可反映肿瘤生长、免疫反应和患者对治疗的反应。在真实研究中,这些标志物会随时间重复测量,但由于技术问题或患者中途退出,常会出现部分读数缺失。简单丢弃不完整记录或用粗略均值填补缺口会严重扭曲结果,尤其当在少量患者中追踪数百种蛋白时。因此,该研究构建了一个按步骤进行的分析流程,旨在尽可能保留信息,同时避免误导性的捷径。
在不“偷看”结局的情况下填补缺失
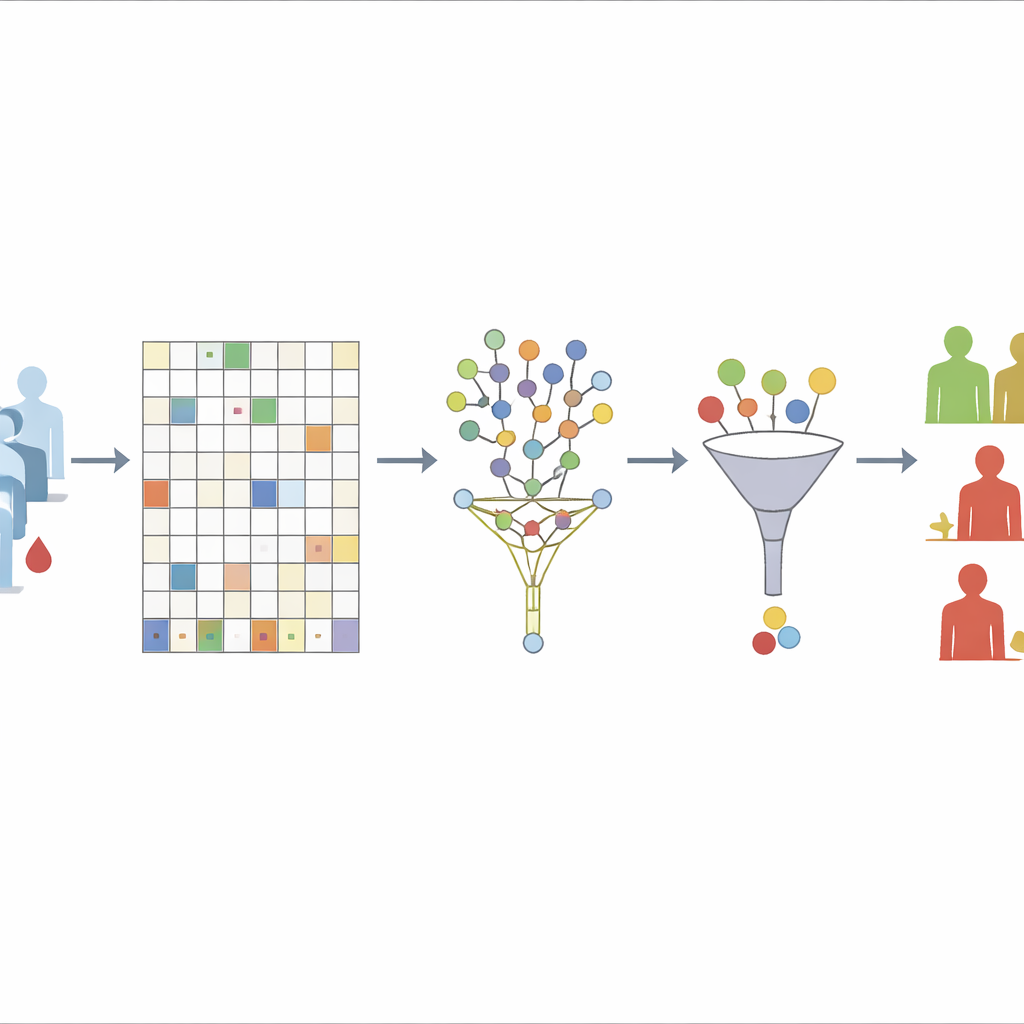
团队首先要解决的挑战是缺失数据。他们仅丢弃在超过30%患者中缺失的蛋白,这一阈值被证明在稳定性和信息保留之间取得了良好平衡。对于其余蛋白,采用一种“无监督”的随机森林方法来推测缺失值。本质上,算法反复构建大量决策树,仅基于蛋白彼此之间的关系,而不考虑谁发生了复发或转移。在许多树中表现相似的样本被视为邻居;然后用这些邻居中已知的数值来填补缺口。通过在此步骤有意排除生存结局,作者避免了在数据清理过程中无意中将答案嵌入数据里的风险。
将数百个标志缩减为有意义的少数
在蛋白表格补全后,下一步是确定哪些标志物确实对预测复发或转移时间有意义。作者首先使用一种将弱预测因子压缩向零而保留强预测因子的方法,实际上像一个筛子,只让最有信息的蛋白通过。鉴于该方法可能遗漏成组相关的标志或细微的非线性模式,他们随后用第二种工具重新审视这些“幸存者”:一种基于为生存数据构建的大量随机决策树的方法。第二阶段并不重新拟合相同的回归模型,而是评估每个标志在树中帮助分裂不同结局患者的频率。那些在树顶端频繁出现的标志被视为更稳定、更重要。
从被选中的标志到患者层面的风险分组
在得到精简的蛋白列表后,作者回到较为传统的生存模型,估计每个标志物及某些临床特征(如肿瘤分期)与无复发或无远处转移概率之间的关系。他们分别为无复发生存和无转移生存建立模型,然后基于患者的蛋白水平和临床特征计算每位患者的风险得分。患者被分为低、中、高风险组,标准的生存曲线显示这些组之间存在明确分离,尽管研究仅包含80名患者。若干蛋白,包括FGF‑5、Neuropilin‑2和与Siglec‑5相关的量度,多次显现为提示较差结局的强指标,而一些标志则表现为保护性因素。
在严苛条件下测试该流程
为了验证他们的方法并非仅对这份小样本数据过拟合,研究者进行了广泛的计算模拟,模拟了在高维蛋白研究中常见的强烈违背常规建模假设的情形以及不同的缺失数据模式。在这些压力测试中,即使经典生存模型背后的常见假设不成立,该流程仍识别出一组紧凑且真正重要的标志,并能区分低风险与高风险组。他们还改变了缺失数据阈值,显示关键标志和总体结论在很大程度上保持稳定。
对未来的意义
这项工作并未发明一种全新的统计技巧,而是组装并验证了一套实用的配方,用以将复杂的蛋白测量转化为临床有意义的风险预测。通过谨慎处理缺失值、聚焦于一组稳定的生物标志物,并通过健壮的内部验证与模拟检查性能,该流程为在小样本、高维的癌症研究中识别有前景的标志物并构建风险评分提供了透明的方法。作者强调,仍需更大且独立的队列来确认某些特定蛋白作为常规临床检测的适用性,但他们的框架为未来以生物标志物为导向的生存研究提供了坚实且可复用的蓝图。
引用: Kumar, N., Bhattacharjee, A., Vishwakarma, G.K. et al. Enhancing survival risk prediction through imputation and feature selection in high-dimensional protein biomarker data. Sci Rep 16, 14490 (2026). https://doi.org/10.1038/s41598-026-43072-z
关键词: 癌症生物标志物, 生存预测, 蛋白质组学, 缺失数据, 精准医学