Clear Sky Science · zh
KinForm:用于酶 kcat 和 KM 预测的动力学知情特征优化表示模型
为什么预测酶速很重要
每个活细胞都依赖酶——这些驱动化学反应的微小蛋白质机器。酶的工作速度以及它们与底物的结合强度,决定了从我们如何代谢食物到微生物如何合成生物燃料的一切过程。在实验室中逐一测量这些动力学特性既缓慢又不完整。本文介绍了 KinForm,这是一种机器学习方法,它从已知样本中学习以估算更多蛋白质的酶学行为,可能帮助生物学家和工程师更有效地设计与分析复杂生化系统。
酶需要告诉我们的信息
理解酶行为时有两个数值特征至关重要。第一个常称为周转数,描述酶在给定时间内可完成多少次反应循环。第二个与酶与底物结合的松紧有关,反映了反应启动的难易。生物学家利用这些数值构建代谢的计算模型,以预测微生物生长或规划新的绿色化学路径。然而,数据库中仅有少部分已知酶具备详尽测量,并且大多数条目集中在少数研究充分的蛋白家族上。这种不均衡的覆盖限制了基础研究和实际应用。
教计算机理解蛋白质的语言
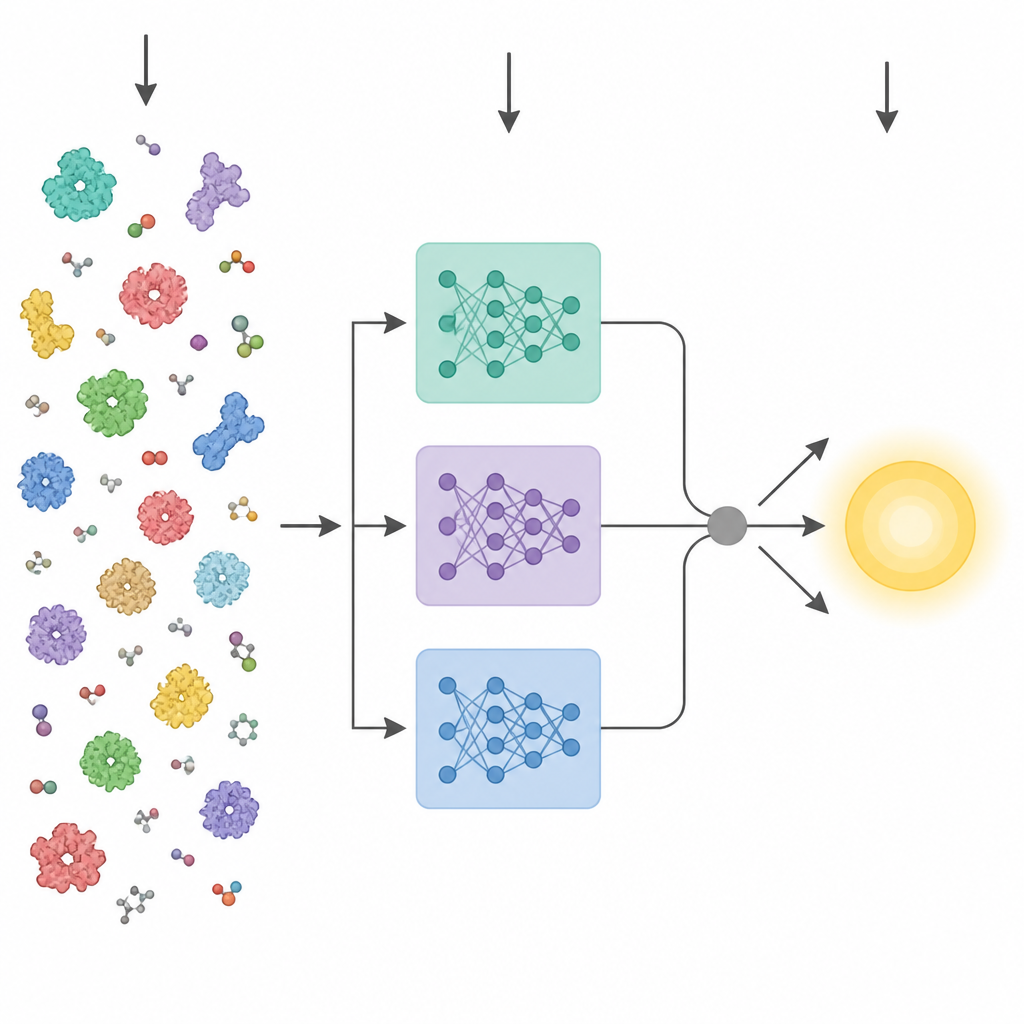
人工智能的最新进展催生了强大的蛋白质“语言模型”。这些工具在数百万条原始氨基酸序列上训练,学习与三维结构和功能相关的模式,而无需实验标注。早期尝试从序列预测酶特性的做法通常将单个模型的最终层压缩为一个平均向量并输入标准预测器。KinForm 走出了一条更为细致的路径。它综合了三种不同的蛋白质语言模型,并关注那些中间层——这些层在动力学预测上比通常使用的最终层携带更多有用信息。
倾听活性位点并去除噪声
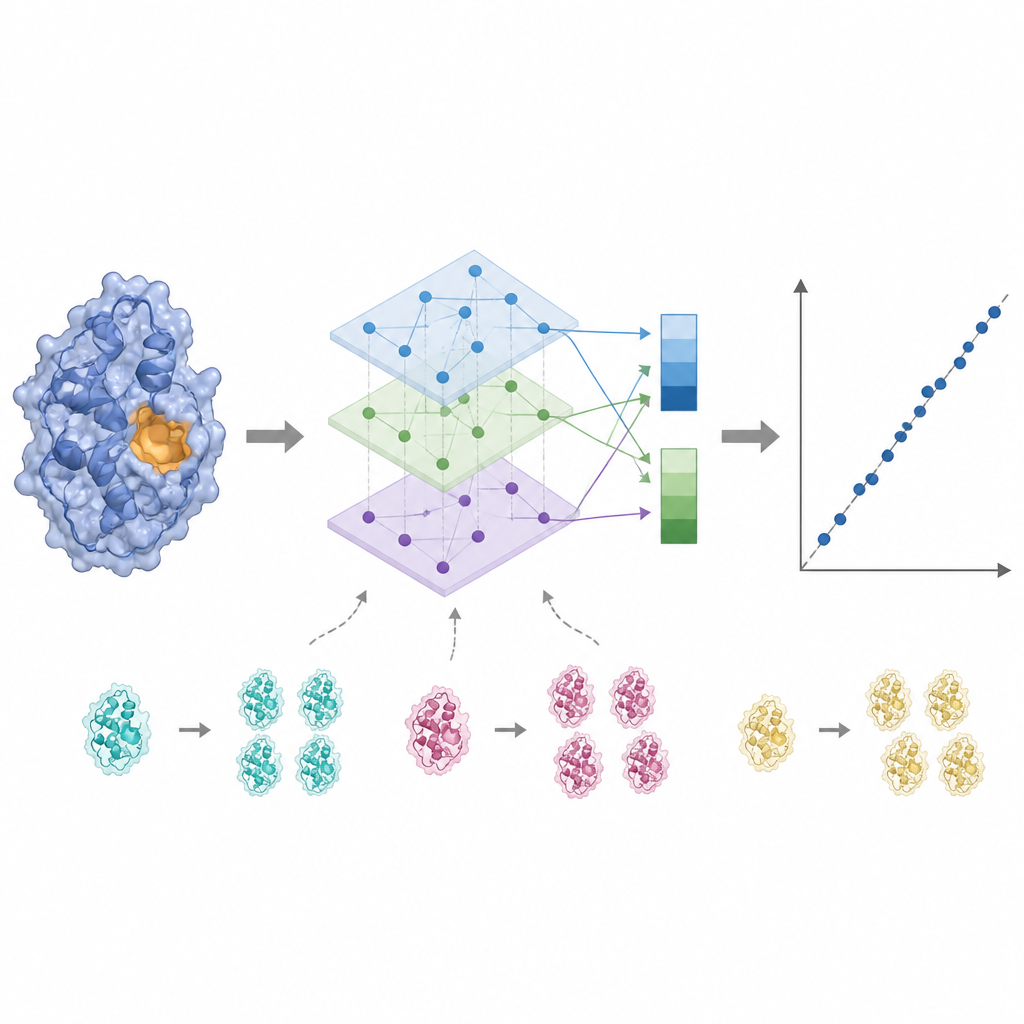
酶活性往往由靠近结合位点的少数关键残基控制,因此把每个氨基酸同等对待会模糊信号。KinForm 通过使用外部工具对每个残基属于结合位点的概率进行评分来纠正这一点。这些评分在对语言模型输出求平均时作为权重,生成一个反映整个蛋白的向量和一个强调预测活性区域的向量。由于以这种方式组合多个大型模型会产生非常高维的数据,KinForm 随后应用主成分分析(一种统计技术),将信息压缩到较少的坐标中,同时保留对预测重要的大部分变异。
处理相似性并测试真正的泛化能力
蛋白质数据库中充斥着密切相关的序列,这可能诱使机器学习模型去记忆而非学习通用规律。KinForm 通过两种方式应对这一问题。首先,在训练过程中故意对那些较罕见、相似性低的蛋白进行过采样,使它们对模型的贡献更大。其次,作者引入了更严格的测试方案,确保训练集与测试集之间不存在任何序列重叠。在这些更苛刻的条件下,使用压缩表示和序列感知采样的 KinForm 变体优于早期方法,尤其是在处理与训练数据差异较大的酶时表现更好。
结果在实践中的意义
在两组大规模酶数据基准上,与现有领先模型相比,KinForm 提高了预测周转数和与结合相关常数的准确性。这些提升在远缘蛋白家族中最为显著——这些家族的数据最稀缺且最需要预测。当将这些预测用于细胞代谢的详细模型时,对整体行为的改进是适度的,表明其他不确定性来源仍然占据重要地位。这项工作表明,精心设计的蛋白质表示和更现实的测试标准可以使基于人工智能的动力学估算更可靠,同时也强调这些估算应被视为不依赖特定条件的起点,而非任何特定环境下的精确数值。
引用: Alwer, S., Fleming, R.M.T. KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction. npj Syst Biol Appl 12, 71 (2026). https://doi.org/10.1038/s41540-026-00692-5
关键词: 酶动力学, 蛋白质语言模型, 机器学习, 代谢建模, 生化预测