Clear Sky Science · en
KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction
Why predicting enzyme speed matters
Every living cell depends on enzymes, tiny protein machines that drive chemical reactions. How fast these enzymes work and how strongly they latch onto their partners shapes everything from how we metabolize food to how microbes make biofuels. Measuring these kinetic traits one enzyme at a time in the lab is slow and incomplete. This article describes KinForm, a machine learning approach that learns from known examples to estimate enzyme behavior for many more proteins, potentially helping biologists and engineers design and analyze complex biochemical systems more effectively.
What enzymes need to tell us
Two numerical traits are central to understanding enzyme behavior. The first, often called the turnover number, describes how many reaction cycles an enzyme can complete in a given time. The second, related to how tightly an enzyme binds its partner molecule, reflects how easily reactions get started. Biologists use these values to build computer models of metabolism, to predict growth of microbes, or to plan new pathways for green chemistry. However, databases contain detailed measurements for only a small share of all known enzymes, and most entries focus on a few well studied protein families. This patchy coverage limits both basic research and practical applications.
Teaching computers the language of proteins
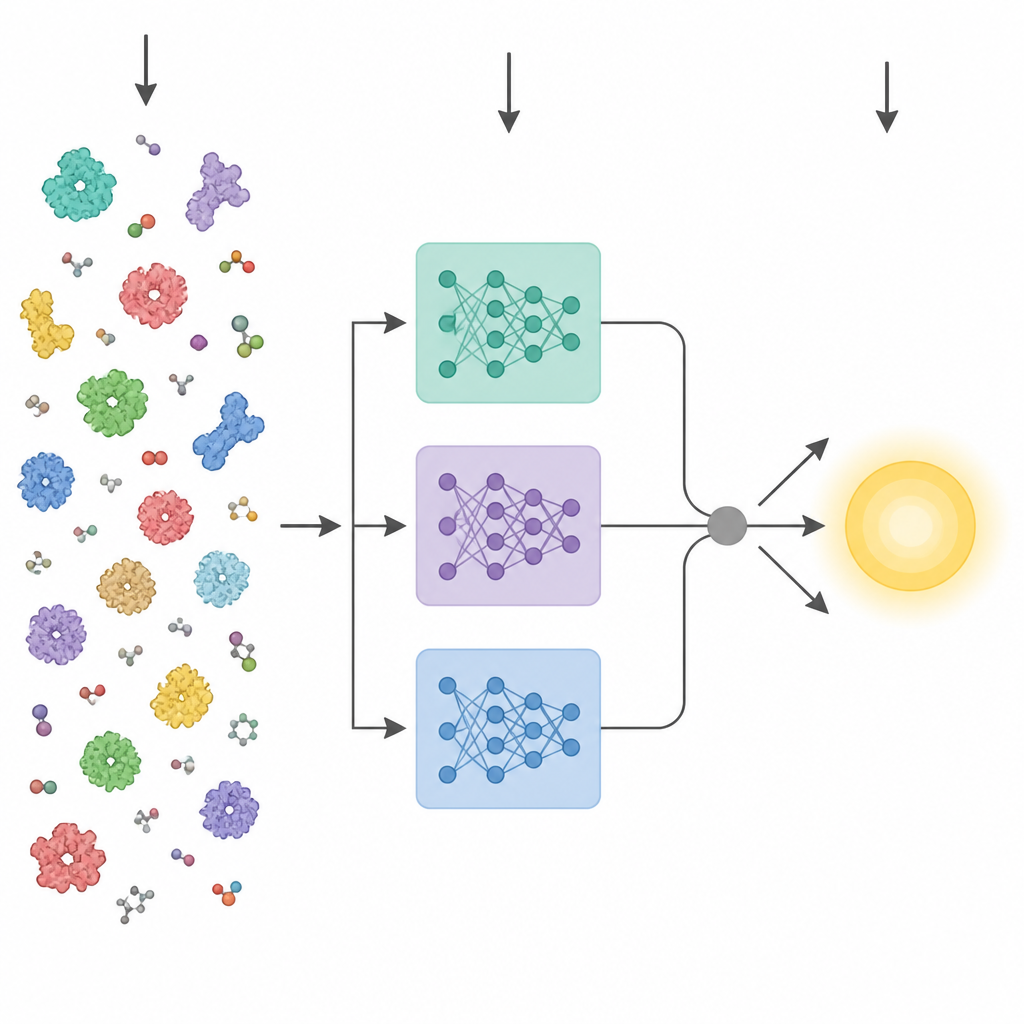
Recent advances in artificial intelligence have produced powerful protein “language models.” These tools are trained on millions of raw amino acid sequences and learn patterns that relate to three dimensional structure and function, without needing any experimental labels. Earlier methods that tried to predict enzyme traits from sequence usually collapsed the final layer of one such model into a single average vector and fed it into a standard predictor. KinForm takes a more nuanced route. It draws on three different protein language models and focuses on intermediate layers that turn out to carry more useful information for kinetic prediction than the usual final layer.
Listening to the active site and trimming the noise
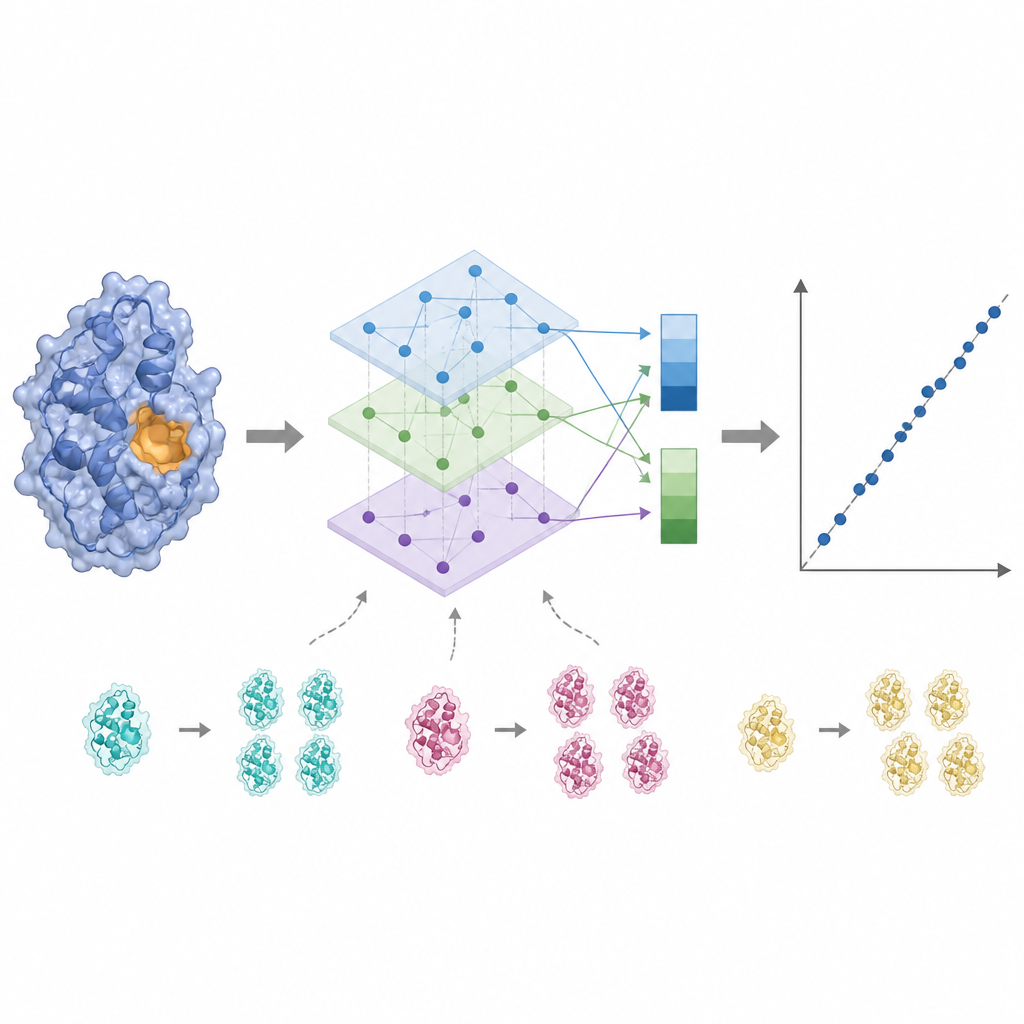
Enzyme activity is often controlled by only a few key residues near the binding site, so treating every amino acid equally can blur the signal. KinForm corrects this by using an external tool that scores how likely each residue is to belong to the binding site. These scores act as weights when averaging the language model outputs, yielding one vector that reflects the whole protein and another that emphasizes the predicted active region. Because combining several large models in this way creates very high dimensional data, KinForm then applies principal component analysis, a statistical technique that compresses the information into a smaller set of coordinates while keeping most of the variation that matters for prediction.
Handling similarity and testing true generalization
Protein databases are crowded with closely related sequences, which can tempt a machine learning model to memorize rather than learn general rules. KinForm tackles this in two ways. First, it deliberately oversamples rarer, low similarity proteins during training so they contribute more strongly to the model. Second, the authors introduce a stricter testing scheme that prevents any overlap in sequence between training and test sets. Under these tougher conditions, KinForm variants that use compressed representations and sequence aware sampling perform better than earlier methods, especially for enzymes that are quite different from anything the model has seen before.
What the results mean in practice
Across two large benchmark collections of enzyme data, KinForm improves the accuracy of predicted turnover numbers and binding related constants compared with a leading prior model. The gains are most striking for distant protein families, where data are scarcest and predictions are most needed. When these predictions are fed into detailed models of cellular metabolism, improvements in overall behavior are modest, suggesting that other sources of uncertainty still play a large role. The work shows that carefully crafted protein representations and more realistic testing standards can make AI based kinetic estimates more reliable, while also highlighting that they should be viewed as condition neutral starting points rather than exact values for any specific environment.
Citation: Alwer, S., Fleming, R.M.T. KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction. npj Syst Biol Appl 12, 71 (2026). https://doi.org/10.1038/s41540-026-00692-5
Keywords: enzyme kinetics, protein language models, machine learning, metabolic modeling, biochemical prediction