Clear Sky Science · fr
KinForm : modèles de représentation optimisés par des caractéristiques informées par la cinétique pour la prédiction de kcat et KM des enzymes
Pourquoi il est important de prédire la vitesse des enzymes
Toute cellule vivante dépend des enzymes, ces petites machines protéiques qui catalysent les réactions chimiques. La vitesse à laquelle ces enzymes agissent et l’affinité avec laquelle elles se lient à leurs partenaires déterminent de nombreux phénomènes, depuis la manière dont nous métabolisons les aliments jusqu’à la production de biocarburants par des micro-organismes. Mesurer ces caractéristiques cinétiques enzyme par enzyme en laboratoire est lent et incomplet. Cet article décrit KinForm, une approche d’apprentissage automatique qui, à partir d’exemples connus, estime le comportement d’un grand nombre de protéines, aidant potentiellement les biologistes et les ingénieurs à concevoir et analyser des systèmes biochimiques complexes de façon plus efficace.
Ce que les enzymes doivent nous dire
Deux grandeurs numériques sont centrales pour comprendre le comportement enzymatique. La première, souvent appelée nombre de rotation (turnover number), décrit combien de cycles réactionnels une enzyme peut accomplir en un temps donné. La seconde, liée à la force de liaison entre l’enzyme et son ligand, reflète la facilité avec laquelle la réaction peut démarrer. Les biologistes utilisent ces valeurs pour construire des modèles informatiques du métabolisme, prédire la croissance microbienne ou planifier de nouvelles voies pour la chimie verte. Cependant, les bases de données ne contiennent des mesures détaillées que pour une petite fraction des enzymes connus, et la plupart des entrées concernent quelques familles protéiques bien étudiées. Cette couverture inégale limite la recherche fondamentale comme les applications pratiques.
Apprendre aux ordinateurs le langage des protéines
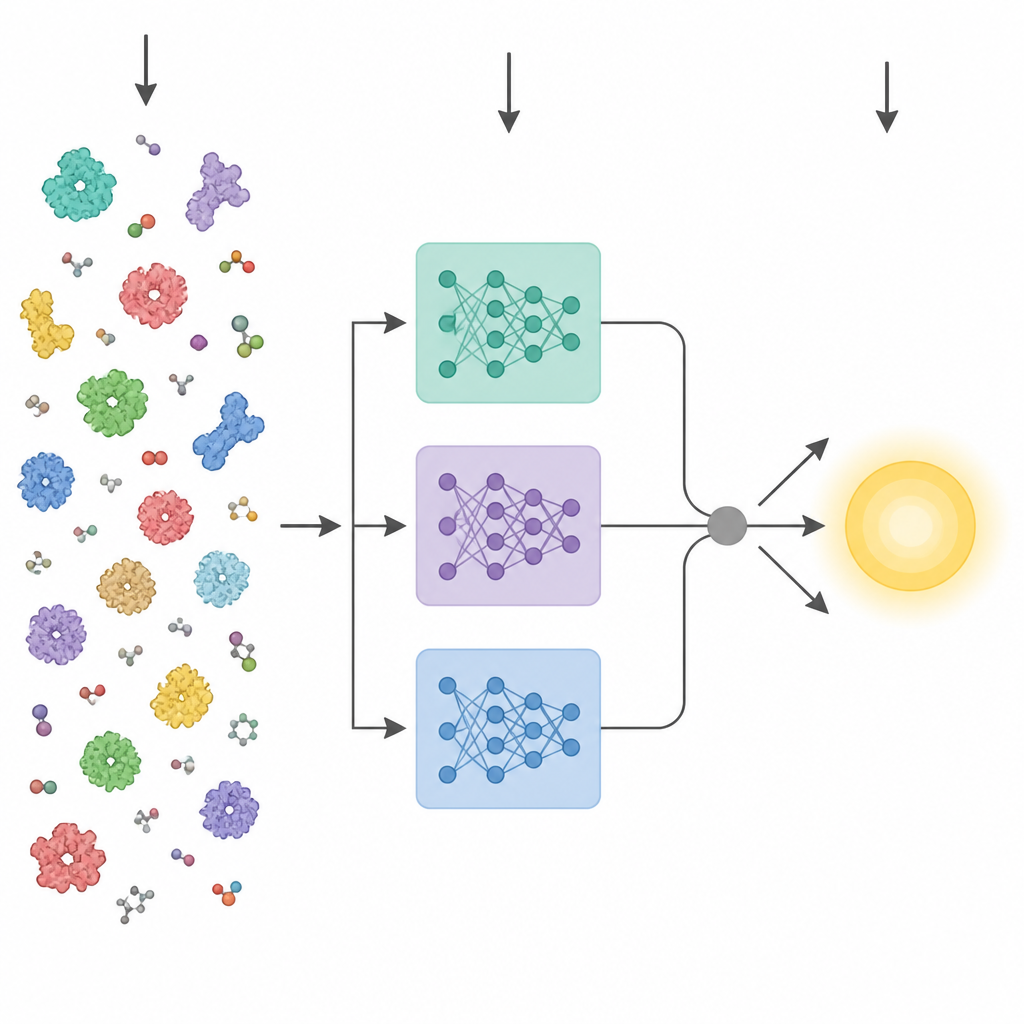
Les progrès récents de l’intelligence artificielle ont donné naissance à de puissants « modèles de langage » protéiques. Ces outils sont entraînés sur des millions de séquences d’acides aminés brutes et apprennent des motifs liés à la structure tridimensionnelle et à la fonction, sans nécessiter d’étiquettes expérimentales. Les méthodes antérieures visant à prédire des propriétés enzymatiques à partir de la séquence compres-saellent généralement la couche finale d’un tel modèle en un vecteur moyen unique puis l’alimentaient à un prédicteur standard. KinForm adopte une approche plus nuancée. Il s’appuie sur trois modèles de langage protéique différents et se concentre sur des couches intermédiaires qui apportent, au final, plus d’informations utiles pour la prédiction cinétique que la couche finale habituelle.
Écouter le site actif et réduire le bruit
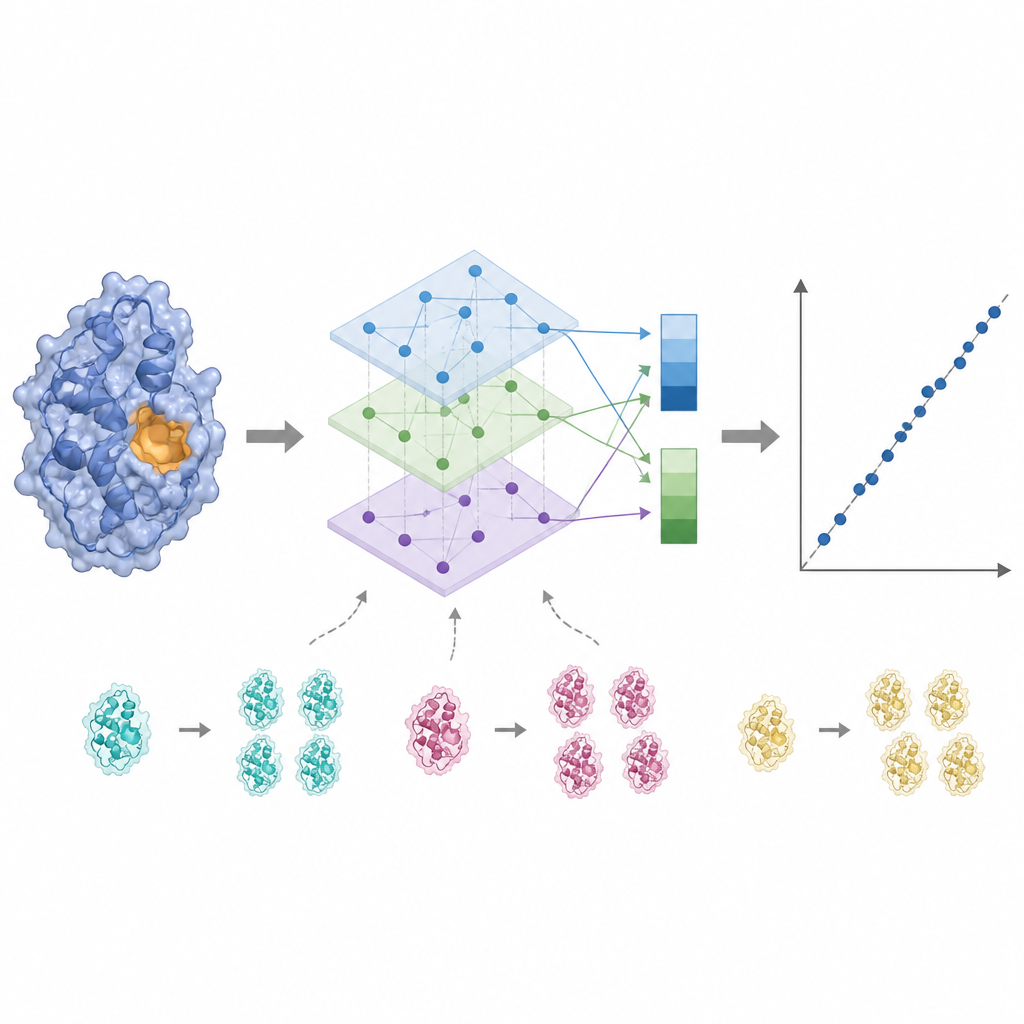
L’activité enzymatique est souvent contrôlée par quelques résidus clés près du site de liaison ; traiter chaque acide aminé de manière égale peut donc diluer le signal. KinForm corrige cela en utilisant un outil externe qui attribue à chaque résidu une probabilité d’appartenir au site de liaison. Ces scores servent de poids lors de la mise en moyenne des sorties des modèles de langage, produisant un vecteur qui reflète la protéine entière et un autre qui met l’accent sur la région active prédite. Parce que la combinaison de plusieurs grands modèles de cette manière génère des données de très haute dimension, KinForm applique ensuite une analyse en composantes principales, une technique statistique qui compresse l’information en un ensemble réduit de coordonnées tout en conservant la majeure partie de la variation pertinente pour la prédiction.
Gérer la similarité et tester la véritable généralisation
Les bases de données protéiques regroupent de nombreuses séquences étroitement reliées, ce qui peut inciter un modèle d’apprentissage automatique à mémoriser plutôt qu’à apprendre des règles générales. KinForm s’attaque à ce problème de deux manières. D’une part, il suréchantillonne délibérément les protéines rares et peu similaires pendant l’entraînement afin qu’elles contribuent davantage au modèle. D’autre part, les auteurs introduisent un schéma de test plus strict qui empêche tout chevauchement de séquences entre les ensembles d’entraînement et de test. Dans ces conditions plus exigeantes, les variantes de KinForm qui utilisent des représentations compressées et un échantillonnage sensible à la séquence surpassent les méthodes précédentes, surtout pour les enzymes assez différentes de tout ce que le modèle a vu auparavant.
Que signifient les résultats en pratique
Sur deux grandes collections de référence de données enzymatiques, KinForm améliore la précision des nombres de rotation prédits et des constantes liées à la liaison par rapport à un modèle de référence antérieur. Les gains sont les plus marqués pour les familles protéiques lointaines, là où les données sont les plus rares et où les prédictions sont les plus nécessaires. Lorsque ces prédictions sont intégrées à des modèles détaillés du métabolisme cellulaire, les améliorations du comportement global restent modestes, ce qui suggère que d’autres sources d’incertitude jouent encore un rôle important. Ce travail montre que des représentations protéiques soigneusement conçues et des standards de test plus réalistes peuvent rendre les estimations cinétiques basées sur l’IA plus fiables, tout en soulignant qu’elles doivent être considérées comme des points de départ neutres vis-à-vis des conditions plutôt que comme des valeurs exactes pour un environnement spécifique.
Citation: Alwer, S., Fleming, R.M.T. KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction. npj Syst Biol Appl 12, 71 (2026). https://doi.org/10.1038/s41540-026-00692-5
Mots-clés: cinétique enzymatique, modèles de langage protéique, apprentissage automatique, modélisation métabolique, prédiction biochimique