Clear Sky Science · es
KinForm: modelos de representación optimizados informados por cinética para la predicción de kcat y KM de enzimas
Por qué importa predecir la velocidad enzimática
Cada célula viva depende de las enzimas, diminunas máquinas proteicas que impulsan las reacciones químicas. La velocidad a la que actúan estas enzimas y la fuerza con la que se unen a sus sustratos condicionan todo, desde cómo metabolizamos los alimentos hasta cómo los microbios producen biocombustibles. Medir estas propiedades cinéticas una enzima a la vez en el laboratorio es lento e incompleto. Este artículo describe KinForm, un enfoque de aprendizaje automático que aprende de ejemplos conocidos para estimar el comportamiento enzimático de muchas más proteínas, ayudando potencialmente a biólogos e ingenieros a diseñar y analizar sistemas bioquímicos complejos con mayor eficacia.
Qué necesitan decirnos las enzimas
Dos magnitudes numéricas son centrales para entender el comportamiento enzimático. La primera, a menudo llamada número de recambio, describe cuántos ciclos de reacción puede completar una enzima en un tiempo dado. La segunda, relacionada con la afinidad de la enzima por su molécula pareja, refleja lo fácil que es iniciar la reacción. Los biólogos usan estos valores para construir modelos computacionales del metabolismo, predecir el crecimiento microbiano o planificar nuevas vías para la química verde. Sin embargo, las bases de datos contienen mediciones detalladas solo para una pequeña fracción de todas las enzimas conocidas, y la mayoría de las entradas se centran en unas pocas familias proteicas bien estudiadas. Esta cobertura fragmentaria limita tanto la investigación básica como las aplicaciones prácticas.
Enseñar a las computadoras el lenguaje de las proteínas
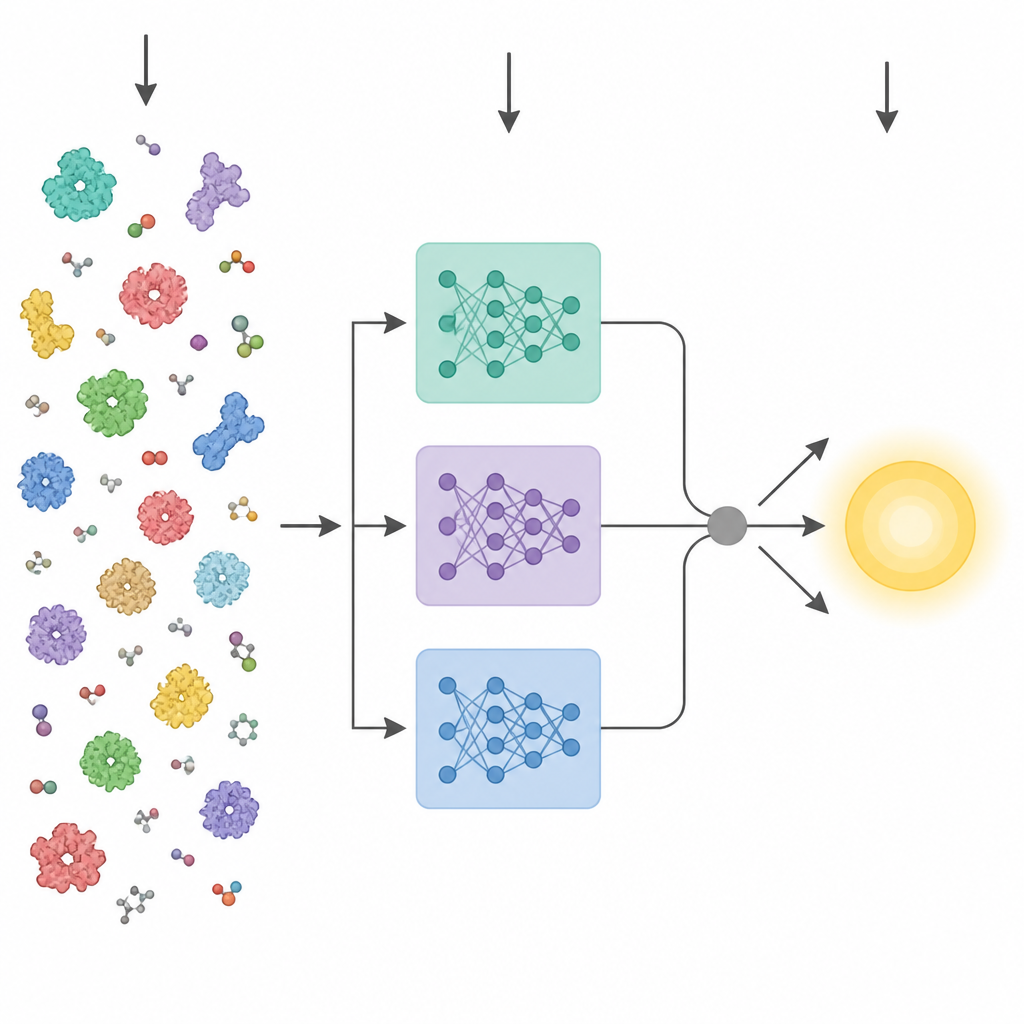
Los avances recientes en inteligencia artificial han producido potentes “modelos de lenguaje” de proteínas. Estas herramientas se entrenan con millones de secuencias de aminoácidos en bruto y aprenden patrones relacionados con la estructura tridimensional y la función, sin necesitar etiquetas experimentales. Los métodos anteriores que intentaron predecir rasgos enzimáticos a partir de la secuencia normalmente colapsaban la capa final de uno de esos modelos en un único vector promedio y lo introducían en un predictor estándar. KinForm sigue una ruta más matizada. Se apoya en tres modelos de lenguaje de proteínas diferentes y se centra en capas intermedias que resultan contener información más útil para la predicción cinética que la típica capa final.
Escuchar el sitio activo y recortar el ruido
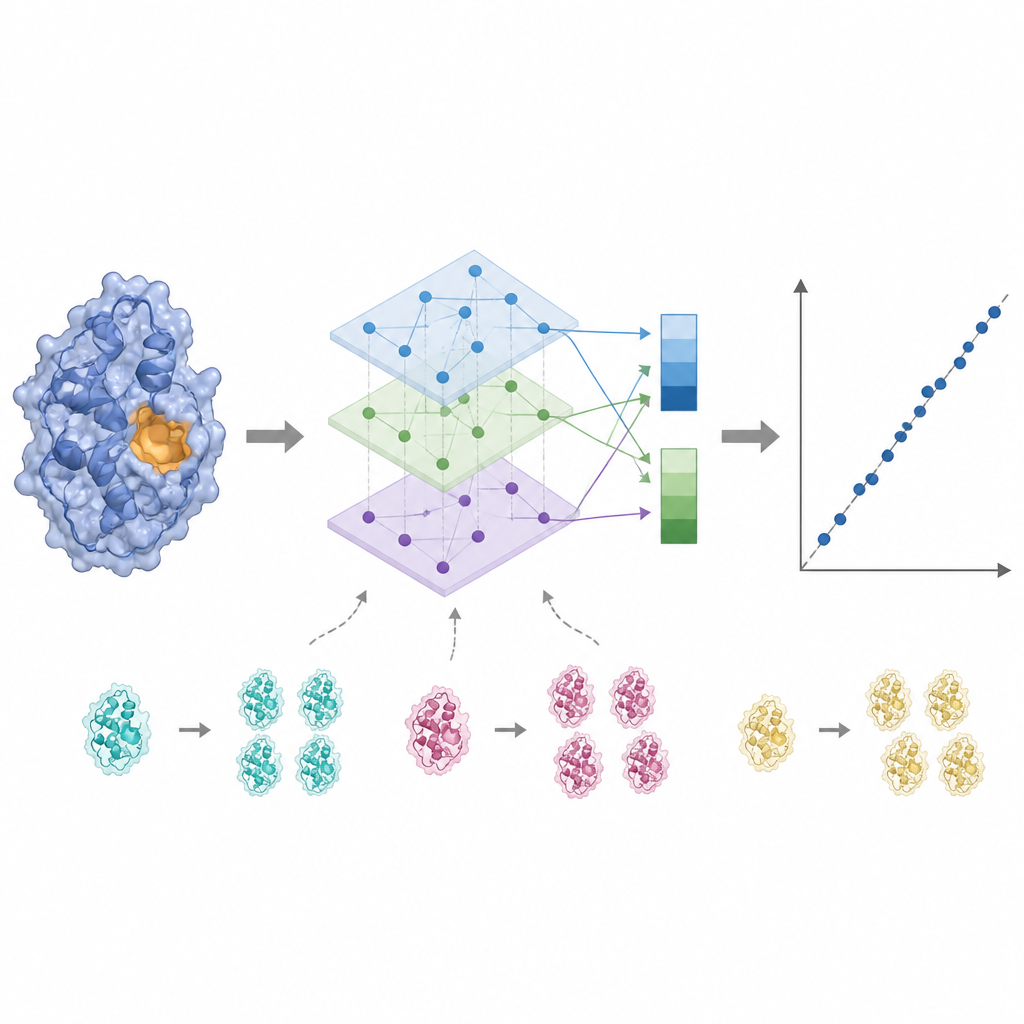
La actividad enzimática suele estar controlada por solo unos pocos residuos clave cercanos al sitio de unión, por lo que tratar cada aminoácido por igual puede difuminar la señal. KinForm corrige esto empleando una herramienta externa que puntúa la probabilidad de que cada residuo pertenezca al sitio de unión. Estas puntuaciones actúan como pesos al promediar las salidas de los modelos de lenguaje, produciendo un vector que refleja la proteína completa y otro que enfatiza la región activa predicha. Dado que combinar varios modelos grandes de esta manera genera datos de muy alta dimensión, KinForm aplica luego análisis de componentes principales, una técnica estadística que comprime la información en un conjunto más pequeño de coordenadas conservando la mayor parte de la variación relevante para la predicción.
Gestionar la similitud y probar la verdadera generalización
Las bases de datos de proteínas están llenas de secuencias estrechamente relacionadas, lo que puede tentar a un modelo de aprendizaje automático a memorizar en lugar de aprender reglas generales. KinForm aborda esto de dos maneras. Primero, sobremuestrea deliberadamente proteínas raras y de baja similitud durante el entrenamiento para que contribuyan con mayor peso al modelo. Segundo, los autores introducen un esquema de prueba más estricto que impide cualquier solapamiento de secuencias entre los conjuntos de entrenamiento y de prueba. Bajo estas condiciones más exigentes, las variantes de KinForm que usan representaciones comprimidas y muestreo sensible a la secuencia rinden mejor que métodos anteriores, sobre todo para enzimas bastante diferentes de cualquier cosa que el modelo haya visto antes.
Qué significan los resultados en la práctica
En dos grandes colecciones de referencia de datos enzimáticos, KinForm mejora la precisión de las predicciones de números de recambio y de constantes relacionadas con la unión en comparación con un modelo previo líder. Las mejoras son más notables en familias proteicas distantes, donde los datos son más escasos y las predicciones más necesarias. Cuando estas predicciones se incorporan en modelos detallados del metabolismo celular, las mejoras en el comportamiento global son modestas, lo que sugiere que otras fuentes de incertidumbre siguen teniendo un papel importante. El trabajo muestra que representaciones proteicas cuidadosamente diseñadas y estándares de prueba más realistas pueden hacer que las estimaciones cinéticas basadas en IA sean más fiables, a la vez que subraya que deben verse como puntos de partida neutrales respecto a condiciones concretas y no como valores exactos para un entorno específico.
Cita: Alwer, S., Fleming, R.M.T. KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction. npj Syst Biol Appl 12, 71 (2026). https://doi.org/10.1038/s41540-026-00692-5
Palabras clave: cinética enzimática, modelos de lenguaje de proteínas, aprendizaje automático, modelado metabólico, predicción bioquímica