Clear Sky Science · pt
KinForm: modelos de representação otimizados e informados por cinética para predição de kcat e KM de enzimas
Por que prever a velocidade de enzimas importa
Cada célula viva depende de enzimas, pequenas máquinas proteicas que impulsionam reações químicas. A velocidade com que essas enzimas atuam e a força com que se ligam a seus parceiros moldam tudo, desde como metabolizamos alimentos até como micróbios produzem biocombustíveis. Medir essas características cinéticas uma enzima por vez no laboratório é lento e incompleto. Este artigo descreve o KinForm, uma abordagem de aprendizado de máquina que aprende com exemplos conhecidos para estimar o comportamento enzimático para muito mais proteínas, potencialmente ajudando biólogos e engenheiros a projetar e analisar sistemas bioquímicos complexos de forma mais eficaz.
O que as enzimas precisam nos dizer
Dois atributos numéricos são centrais para entender o comportamento enzimático. O primeiro, frequentemente chamado de número de turnover, descreve quantos ciclos de reação uma enzima pode completar em um dado tempo. O segundo, relacionado a quão firmemente a enzima se liga à sua molécula parceira, reflete com que facilidade as reações se iniciam. Biólogos usam esses valores para construir modelos computacionais do metabolismo, prever crescimento de micróbios ou planejar novas vias para química verde. Entretanto, bancos de dados contêm medições detalhadas para apenas uma pequena parcela de todas as enzimas conhecidas, e a maioria das entradas foca em algumas famílias proteicas bem estudadas. Essa cobertura fragmentada limita tanto a pesquisa básica quanto aplicações práticas.
Ensinando computadores a linguagem das proteínas
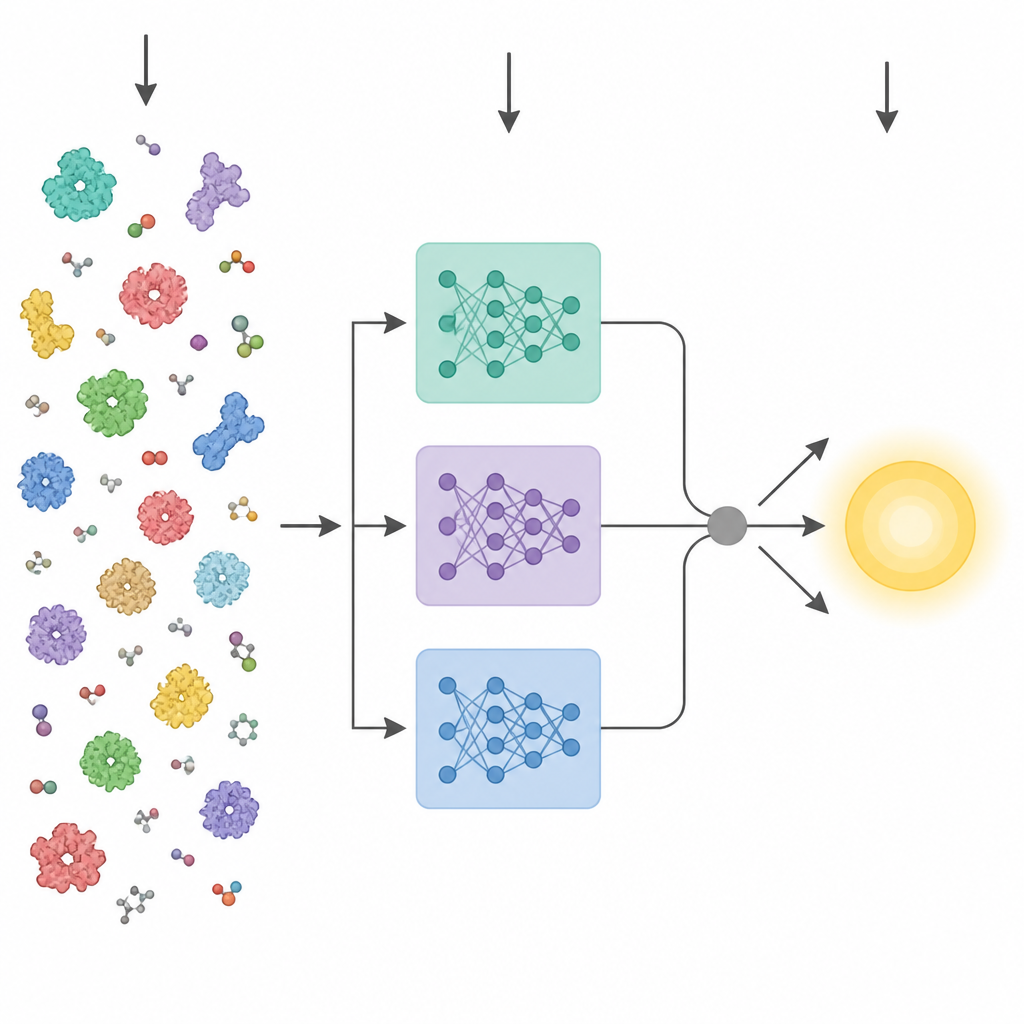
Avanços recentes em inteligência artificial produziram poderosos “modelos de linguagem” para proteínas. Essas ferramentas são treinadas em milhões de sequências de aminoácidos brutas e aprendem padrões relacionados à estrutura tridimensional e à função, sem precisar de rótulos experimentais. Métodos anteriores que tentaram predizer características enzimáticas a partir da sequência geralmente colapsavam a camada final de um desses modelos em um único vetor médio e o alimentavam em um preditor padrão. O KinForm segue um caminho mais nuançado. Ele recorre a três modelos de linguagem de proteínas diferentes e foca em camadas intermediárias que se mostram mais informativas para a predição cinética do que a camada final habitual.
Ouvindo o sítio ativo e reduzindo o ruído
A atividade enzimática costuma ser controlada por apenas alguns resíduos-chave próximos ao sítio de ligação, então tratar cada aminoácido igualmente pode borrar o sinal. O KinForm corrige isso usando uma ferramenta externa que pontua quão provável cada resíduo é de pertencer ao sítio de ligação. Essas pontuações atuam como pesos ao se fazer a média das saídas dos modelos de linguagem, gerando um vetor que reflete a proteína inteira e outro que enfatiza a região ativa prevista. Como combinar vários modelos grandes dessa forma cria dados de dimensionalidade muito alta, o KinForm então aplica análise de componentes principais, uma técnica estatística que comprime a informação em um conjunto menor de coordenadas enquanto preserva a maior parte da variação relevante para predição.
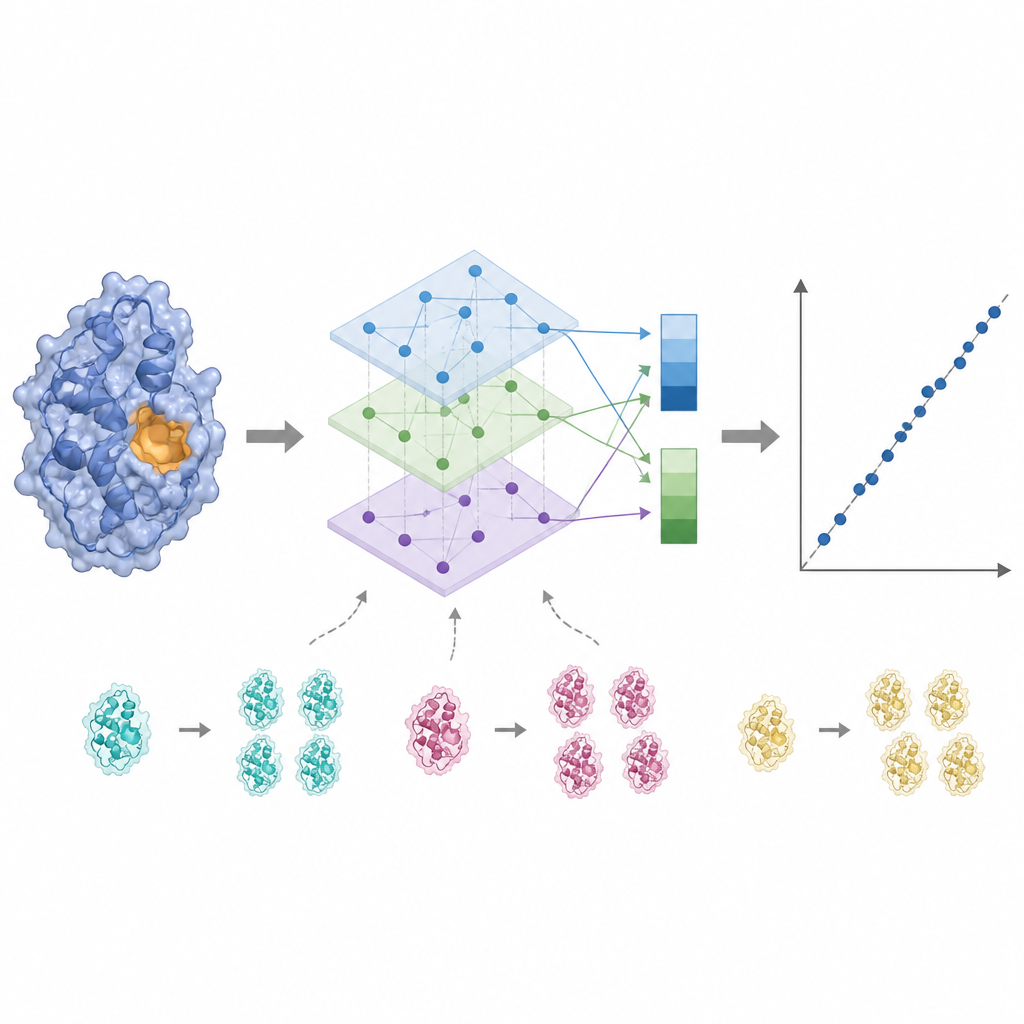
Lidando com similaridade e testando a verdadeira generalização
Bancos de dados de proteínas estão cheios de sequências estreitamente relacionadas, o que pode levar um modelo de aprendizado de máquina a memorizar em vez de aprender regras gerais. O KinForm enfrenta isso de duas maneiras. Primeiro, ele sobremostra deliberadamente proteínas mais raras e de baixa similaridade durante o treinamento para que elas contribuam mais fortemente ao modelo. Segundo, os autores introduzem um esquema de teste mais rigoroso que impede qualquer sobreposição de sequência entre conjuntos de treinamento e teste. Nestas condições mais duras, variantes do KinForm que usam representações comprimidas e amostragem consciente da sequência têm desempenho melhor que métodos anteriores, especialmente para enzimas bastante diferentes de qualquer coisa que o modelo já tenha visto.
O que os resultados significam na prática
Em duas grandes coleções de referência de dados enzimáticos, o KinForm melhora a precisão das predições de números de turnover e constantes relacionadas à ligação quando comparado a um modelo anterior de referência. Os ganhos são mais notórios para famílias de proteínas distantes, onde os dados são mais escassos e as predições mais necessárias. Quando essas predições são inseridas em modelos detalhados do metabolismo celular, as melhorias no comportamento geral são modestas, o que sugere que outras fontes de incerteza ainda exercem grande influência. O trabalho mostra que representações proteicas cuidadosamente elaboradas e padrões de teste mais realistas podem tornar estimativas cinéticas baseadas em IA mais confiáveis, ao mesmo tempo em que ressalta que elas devem ser vistas como pontos de partida neutros em relação às condições, e não como valores exatos para um ambiente específico.
Citação: Alwer, S., Fleming, R.M.T. KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction. npj Syst Biol Appl 12, 71 (2026). https://doi.org/10.1038/s41540-026-00692-5
Palavras-chave: cinética enzimática, modelos de linguagem de proteínas, aprendizado de máquina, modelagem metabólica, predição bioquímica