Clear Sky Science · sv
KinForm: kinetik‑informerade funktionsoptimerade representationsmodeller för prediktion av enzymets kcat och KM
Varför det är viktigt att förutsäga enzymhastighet
Varje levande cell är beroende av enzymer, små proteinmaskiner som driver kemiska reaktioner. Hur snabbt dessa enzymer arbetar och hur starkt de binder sina partners formar allt från hur vi metaboliserar mat till hur mikrober producerar biobränslen. Att mäta dessa kinetiska egenskaper ett enzym i taget i labbet är långsamt och ofullständigt. Denna artikel beskriver KinForm, ett maskininlärningssätt som lär sig från kända exempel för att uppskatta enzymbeteende för många fler proteiner, vilket potentiellt hjälper biologer och ingenjörer att bättre designa och analysera komplexa biokemiska system.
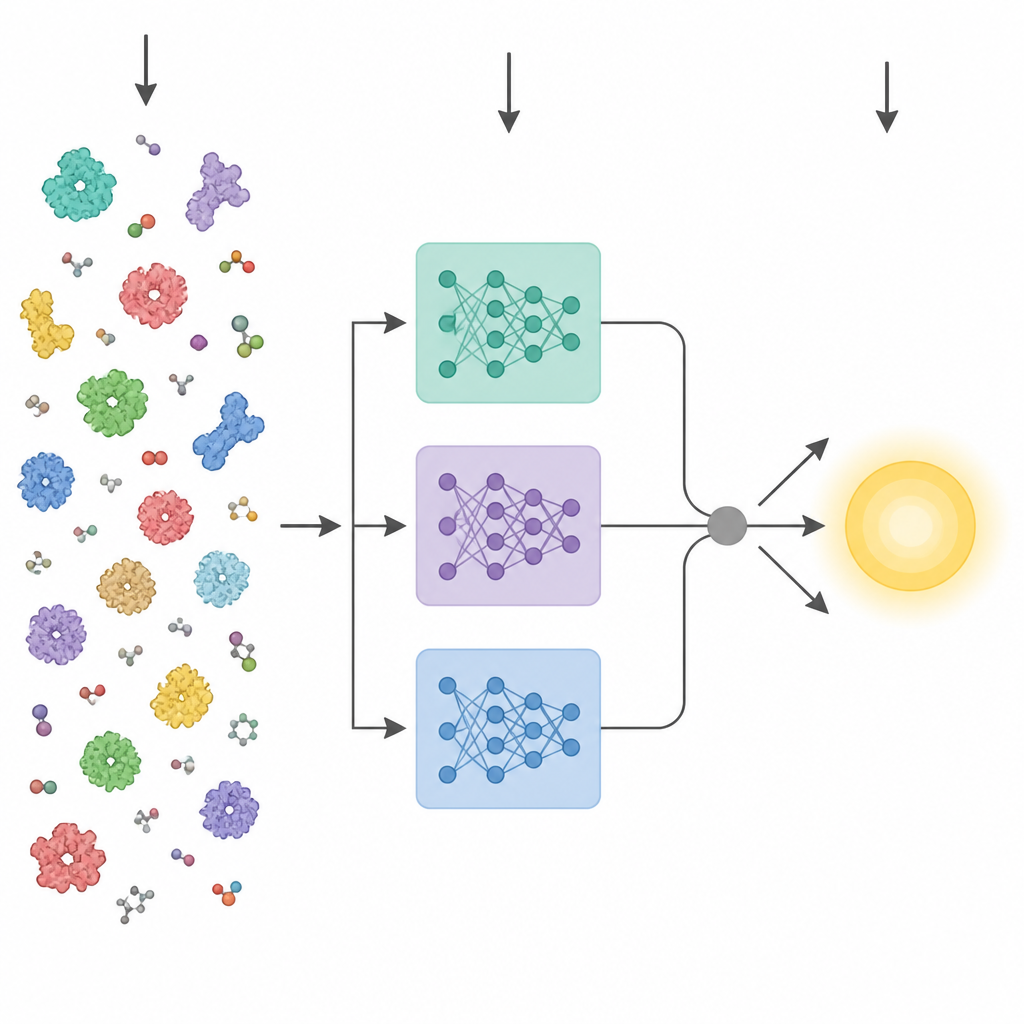
Vad enzymer måste berätta för oss
Två numeriska egenskaper är centrala för att förstå enzymbeteende. Den första, ofta kallad turnover‑nummer, beskriver hur många reaktionscykler ett enzym kan genomföra under en given tid. Den andra, relaterad till hur tätt ett enzym binder sitt substrat, speglar hur lätt reaktionerna kommer igång. Biologer använder dessa värden för att bygga datormodeller av metabolism, för att förutsäga mikrobers tillväxt eller för att planera nya vägar för grön kemi. Databaser innehåller dock detaljerade mätningar för endast en liten andel av alla kända enzymer, och de flesta poster fokuserar på ett fåtal välstuderade proteinfamiljer. Denna ojämna täckning begränsar både grundforskning och praktiska tillämpningar.
Att lära datorer proteiners språk
Nyliga framsteg inom artificiell intelligens har gett kraftfulla protein‑”språkmodeller.” Dessa verktyg tränas på miljontals råa aminosyrasekvenser och lär sig mönster som relaterar till tredimensionell struktur och funktion, utan att behöva experimentella etiketter. Tidigare metoder som försökte förutsäga enzymegenskaper från sekvens brukade ofta komprimera det sista lagret i en sådan modell till en enda genomsnittlig vektor och mata in den i en standardprediktor. KinForm väljer en mer nyanserad väg. Den hämtar information från tre olika protein‑språkmodeller och fokuserar på mellanliggande lager som visar sig bära mer användbar information för kinetisk prediktion än det vanliga slutlagret.
Lyssna på det aktiva sätet och ta bort brus
Enzymaktivitet styrs ofta av bara några nyckelresiduier nära bindningsstället, så att behandla varje aminosyra lika kan sudda ut signalen. KinForm åtgärdar detta genom att använda ett externt verktyg som poängsätter hur sannolikt varje residu tillhör bindningsstället. Dessa poäng fungerar som vikter vid medelvärdesbildning av språkmodellernas output, vilket ger en vektor som speglar hela proteinet och en annan som betonar den förutspådda aktiva regionen. Eftersom kombinationen av flera stora modeller på detta sätt skapar mycket högdimensionell data, applicerar KinForm sedan principal component analysis, en statistisk teknik som komprimerar informationen till ett mindre antal koordinater samtidigt som den behåller det mesta av den variation som är relevant för prediktion.
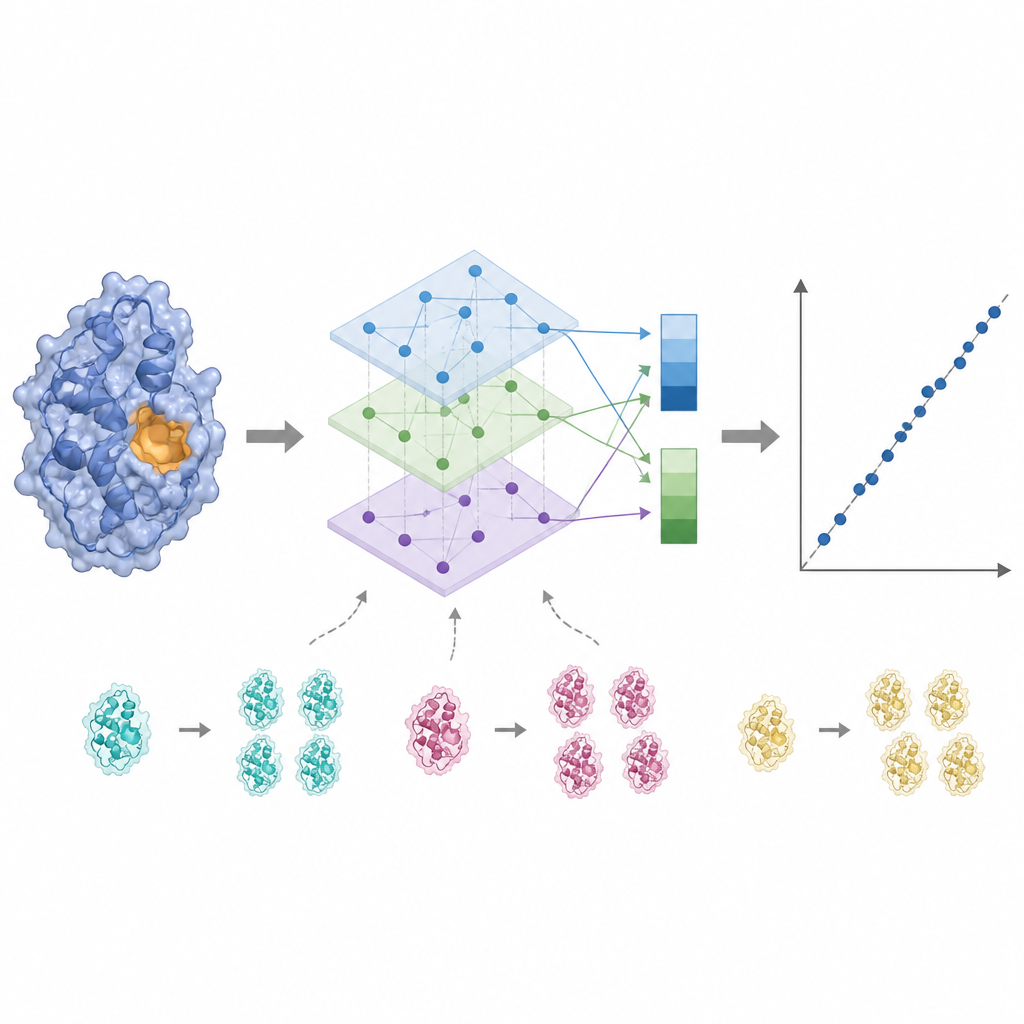
Hantera likhet och testa verklig generalisering
Proteindatabaser är fulla av nära besläktade sekvenser, vilket kan fresta en maskininlärningsmodell att memorera snarare än lära generella regler. KinForm tacklar detta på två sätt. För det första översampelar den medvetet sällsyntare, låg‑likhetsproteiner under träningen så att de bidrar starkare till modellen. För det andra introducerar författarna en striktare testprocedur som förhindrar någon överlappning i sekvens mellan tränings‑ och testuppsättningar. Under dessa tuffare villkor presterar KinForm‑varianter som använder komprimerade representationer och sekvensmedveten sampning bättre än tidigare metoder, särskilt för enzymer som är ganska olika allt modellen tidigare sett.
Vad resultaten betyder i praktiken
Över två stora referenssamlingar av enzymdata förbättrar KinForm noggrannheten i predikterade turnover‑nummer och bindningsrelaterade konstanter jämfört med en ledande tidigare modell. Vinsterna är mest påtagliga för avlägsna proteinfamiljer, där data är knappast och prediktioner är som mest efterfrågade. När dessa prediktioner matas in i detaljerade modeller av cellulär metabolism är förbättringarna i det övergripande beteendet modesta, vilket tyder på att andra osäkerhetskällor fortfarande spelar en stor roll. Studien visar att noggrant utformade proteinrepresentationer och mer realistiska teststandarder kan göra AI‑baserade kinetiska uppskattningar mer tillförlitliga, samtidigt som den understryker att de bör ses som tillståndsneutrala utgångspunkter snarare än exakta värden för en specifik miljö.
Citering: Alwer, S., Fleming, R.M.T. KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction. npj Syst Biol Appl 12, 71 (2026). https://doi.org/10.1038/s41540-026-00692-5
Nyckelord: enzymkinetik, protein‑språkmodeller, maskininlärning, metabol modellering, biokemisk prediktion