Clear Sky Science · it
KinForm: modelli di rappresentazione ottimizzati con informazioni cinetiche per la previsione di kcat e KM degli enzimi
Perché è importante prevedere la velocità degli enzimi
Ogni cellula vivente dipende dagli enzimi, piccole macchine proteiche che guidano le reazioni chimiche. La velocità con cui questi enzimi operano e quanto fortemente si legano ai loro partner determina tutto, dal modo in cui metabolizziamo il cibo a come i microrganismi producono biocarburanti. Misurare questi tratti cinetici un enzima alla volta in laboratorio è lento e incompleto. Questo articolo descrive KinForm, un approccio di apprendimento automatico che impara da esempi noti per stimare il comportamento enzimatico su molti più proteine, aiutando potenzialmente biologi e ingegneri a progettare e analizzare sistemi biochimici complessi in modo più efficace.
Cosa devono dirci gli enzimi
Due caratteristiche numeriche sono centrali per comprendere il comportamento enzimatica. La prima, spesso chiamata numero di turnover, descrive quante cicli reazionali un enzima può completare in un dato intervallo di tempo. La seconda, relativa a quanto saldamente un enzima si lega alla sua molecola partner, riflette quanto facilmente si avviano le reazioni. I biologi usano questi valori per costruire modelli computazionali del metabolismo, per prevedere la crescita dei microrganismi o per progettare nuovi percorsi per la chimica sostenibile. Tuttavia, i database contengono misurazioni dettagliate solo per una piccola parte di tutti gli enzimi conosciuti, e la maggior parte delle voci si concentra su poche famiglie proteiche ben studiate. Questa copertura frammentaria limita sia la ricerca di base sia le applicazioni pratiche.
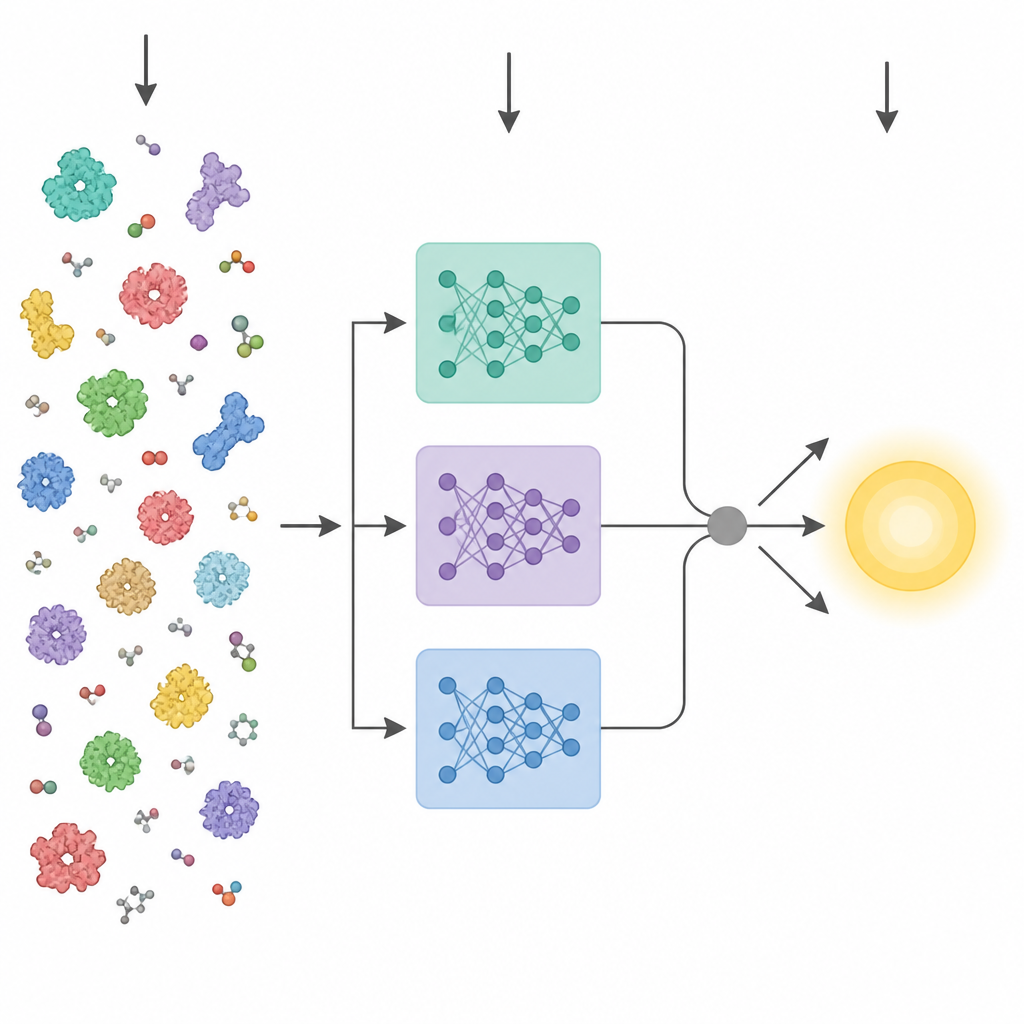
Insegnare ai computer il linguaggio delle proteine
I recenti progressi nell’intelligenza artificiale hanno prodotto potenti “modelli linguistici” per le proteine. Questi strumenti sono addestrati su milioni di sequenze di amminoacidi grezze e apprendono pattern che si correlano con la struttura tridimensionale e la funzione, senza bisogno di etichette sperimentali. I metodi precedenti che cercavano di prevedere i tratti enzimatici dalla sequenza solitamente comprimono lo strato finale di uno di questi modelli in un singolo vettore medio e lo inviano a un predittore standard. KinForm adotta un percorso più sfumato. Si avvale di tre diversi modelli linguistici per proteine e si concentra su strati intermedi che risultano contenere informazioni più utili per la previsione cinetica rispetto al consueto strato finale.
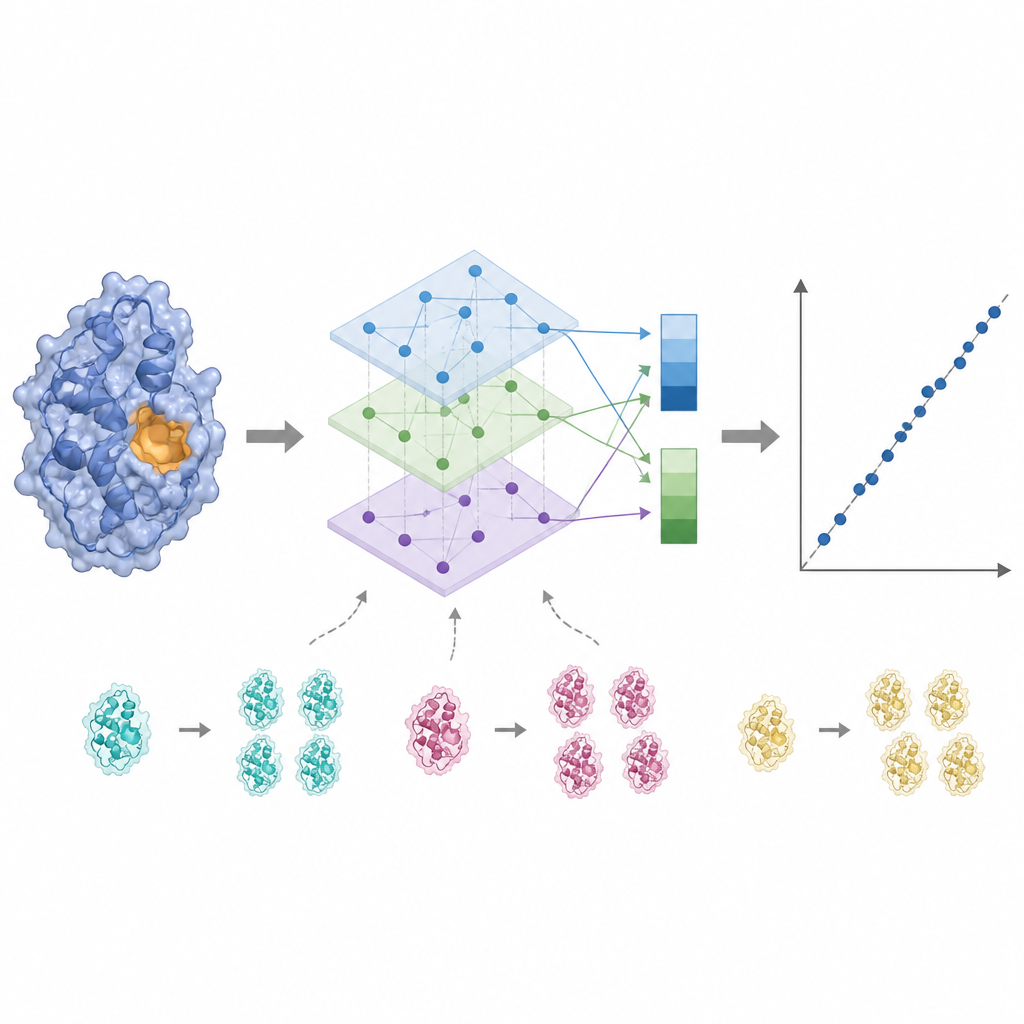
Ascoltare il sito attivo e ridurre il rumore
L’attività enzimatica è spesso determinata da poche residui chiave vicine al sito di legame, quindi trattare ogni amminoacido allo stesso modo può offuscare il segnale. KinForm corregge questo usando uno strumento esterno che assegna un punteggio alla probabilità che ciascun residuo appartenga al sito di legame. Questi punteggi fungono da pesi quando si mediano gli output dei modelli linguistici, generando un vettore che riflette l’intera proteina e un altro che enfatizza la regione attiva prevista. Poiché combinare diversi grandi modelli in questo modo crea dati ad alta dimensionalità, KinForm applica poi l’analisi delle componenti principali, una tecnica statistica che comprime l’informazione in un set più piccolo di coordinate mantenendo la maggior parte della variazione rilevante per la predizione.
Gestire la somiglianza e testare la vera generalizzazione
I database di proteine sono affollati di sequenze strettamente correlate, il che può indurre un modello di apprendimento automatico a memorizzare invece che apprendere regole generali. KinForm affronta questo problema in due modi. Primo, sovracampiona deliberatamente le proteine più rare e a bassa somiglianza durante l’addestramento in modo che contribuiscano più fortemente al modello. Secondo, gli autori introducono uno schema di test più rigoroso che impedisce qualsiasi sovrapposizione di sequenze tra i set di addestramento e di test. In queste condizioni più severe, le varianti di KinForm che usano rappresentazioni compresse e campionamento consapevole della sequenza ottengono prestazioni migliori rispetto ai metodi precedenti, soprattutto per enzimi piuttosto diversi da tutto ciò che il modello ha già visto.
Cosa significano i risultati nella pratica
Su due grandi collezioni di benchmark di dati enzimatici, KinForm migliora l’accuratezza della previsione dei numeri di turnover e delle costanti correlate al legame rispetto a un modello precedente di riferimento. I guadagni sono più evidenti per famiglie proteiche distanti, dove i dati sono più scarsi e le previsioni più necessarie. Quando queste previsioni vengono incorporate in modelli dettagliati del metabolismo cellulare, i miglioramenti nel comportamento complessivo sono modesti, suggerendo che altre fonti di incertezza giocano ancora un ruolo importante. Il lavoro dimostra che rappresentazioni proteiche curate e standard di test più realistici possono rendere le stime cinetiche basate sull’IA più affidabili, evidenziando al contempo che dovrebbero essere considerate punti di partenza neutrali rispetto alle condizioni piuttosto che valori esatti per uno specifico ambiente.
Citazione: Alwer, S., Fleming, R.M.T. KinForm: kinetics-informed feature optimised representation models for enzyme kcat and KM prediction. npj Syst Biol Appl 12, 71 (2026). https://doi.org/10.1038/s41540-026-00692-5
Parole chiave: cinetica enzimatica, modelli linguistici per proteine, apprendimento automatico, modellazione metabolica, predizione biochimica