Clear Sky Science · zh
用最少的从头算数据构建机器学习原子间势
更聪明的模拟,造就更好的电池
固态电池通过用能传导锂离子的固体材料替代易燃的液体电解质,有望使手机、汽车和电网储能更安全。但寻找和测试新的固体导体既缓慢又昂贵,尤其是当研究人员依赖跟踪每个电子的昂贵超算计算时。本文展示了如何利用现代机器学习大幅降低这一成本:作者仅用几百次昂贵计算而非数万次,就构建出准确且快速的原子力“数字孪生体”,从而为下一代电池材料的快速筛选打开了大门。
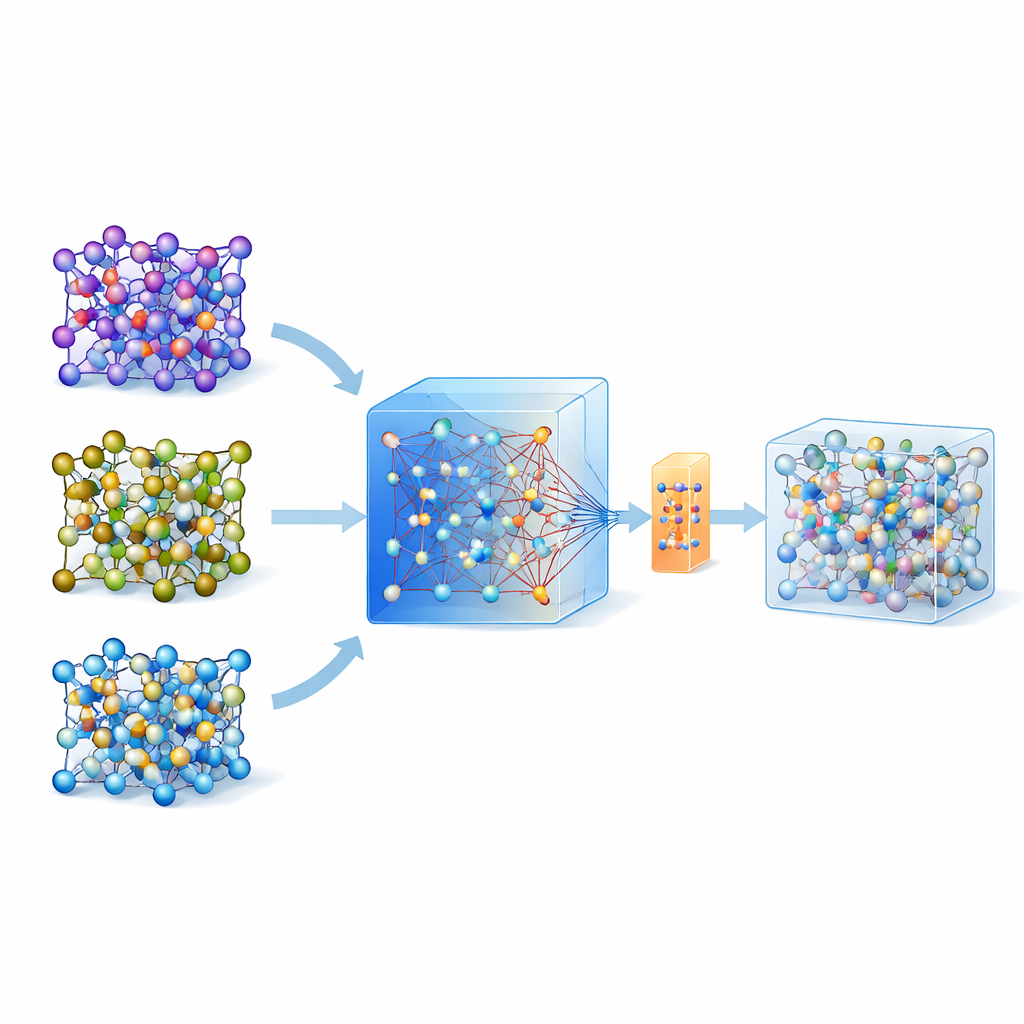
为何原子模拟如此困难
为了判断一种固体材料能否快速传导锂离子,科学家常常求助于从头算分子动力学(ab initio molecular dynamics),这是一种基于量子力学计算原子运动的黄金标准。问题在于它计算量极大,无法常规用于大尺度体系或长时间演化。机器学习原子间势提供了一条捷径:一旦训练完成,它们能以极低的开销模拟底层的量子力。然而,为特定材料构建此类模型传统上需要复杂的“主动学习”循环和成千上万次量子计算,这大大限制了它们的广泛应用。
以大型通用模型为向导
近年出现了在跨材料大数据库上训练的通用大型机器学习模型。其中一个名为 MACE-MP-0 的模型就是本文的起点。作者首先在三种具有代表性的固态电解质上测试了该通用模型,覆盖不同化学体系:硫化物(LGPS)、氧化物(LATP)和卤化物(Li3YCl6)。结果表明,虽然 MACE-MP-0 能大致再现昂贵参考模拟的原子轨迹,但在预测诸如锂迁移势垒和扩散速率等精细性质上还不够精确。尽管如此,它在原子构型空间中的运动与高精度计算高度一致,使其成为一个出色且廉价的相关原子结构“采样器”。
从极小数据集构建精确模型
作者提出了一个非反复更新的单次策略:首先使用通用 MACE 模型在高温下进行分子动力学以生成大量原子快照,然后采用智能重采样方法挑选出大约 200 个特别有信息量的构型,并用完整的量子方法计算它们的能量与力。作者没有从头训练一个新模型,而是在这个小而精心挑选的数据集上微调已有的 MACE 模型,既采用常规微调,也使用一种参数高效的变体 ELoRA。经过微调的模型不仅在能垒和扩散等方面显著提高了精度,还继承了原始大模型的动力学稳定性,避免了在数据极少时常见的非物理性原子坍缩问题。
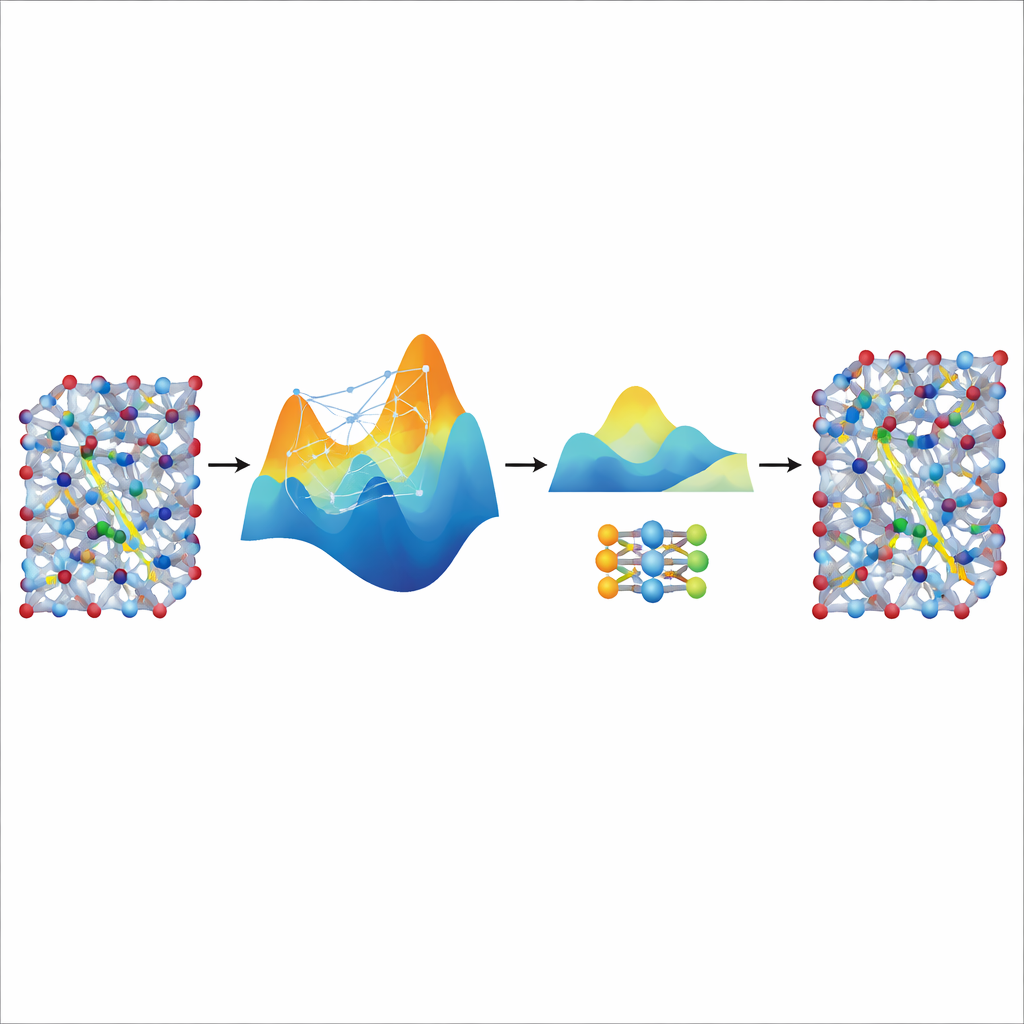
把大型教师的速度蒸馏出来
尽管微调后的 MACE 模型准确且稳定,但对于需要研究真实电池材料中离子传输的超长和大尺度模拟来说,它仍然相对笨重且较慢。为此,作者将其作为“教师”来训练一个更小、更轻量的学生模型 NEP。他们让微调后的 MACE 模型生成额外的合成训练数据——成千上万个带有其预测能量和力的原子构型——无需额外的量子计算。在这份蒸馏数据上训练 NEP 后,得到的紧凑模型运行速度提升约二十倍,同时在很大程度上匹配教师的预测。在大超胞模拟中,蒸馏得到的 NEP 模型能再现诸如超离子相变和室温电导率等关键特征,并与实验结果良好一致。
对未来材料研究的意义
这项研究展示了一套实用的配方:只用几百次昂贵的量子计算,就能构建出可靠且快速的机器学习力场——先用通用模型广泛采样,精细微调,再将其知识蒸馏到更精简的学生模型上。对于固态电解质,这一方法使得长时程、大尺度模拟成为可能,能够直接捕捉锂离子在复杂晶格中穿梭的真实行为,给出更接近实际的电导率而非粗略估计。更广泛地说,同样的工作流程可以加速多种功能性材料的设计,使例行化、高保真原子尺度模拟更接近日常研究实践。
引用: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
关键词: 固态电解质, 机器学习势, 分子动力学, 电池材料, 材料模拟