Clear Sky Science · it
Costruire potenziali interatomici di machine learning con la minima quantità di dati ab initio
Simulazioni più intelligenti per batterie migliori
Le batterie a stato solido promettono telefoni, auto e accumulo di rete più sicuri sostituendo elettroliti liquidi infiammabili con materiali solidi che conducono gli ioni di litio. Ma trovare e testare nuovi conduttori solidi è lento e costoso, soprattutto quando i ricercatori si affidano a pesanti calcoli su supercomputer che seguono ogni elettrone. Questo articolo mostra come usare il machine learning moderno per ridurre drasticamente questo costo: gli autori costruiscono “gemelli digitali” delle forze atomiche precisi e rapidi usando solo poche centinaia di calcoli costosi invece di decine di migliaia, aprendo la strada allo screening rapido di materiali per batterie di nuova generazione.
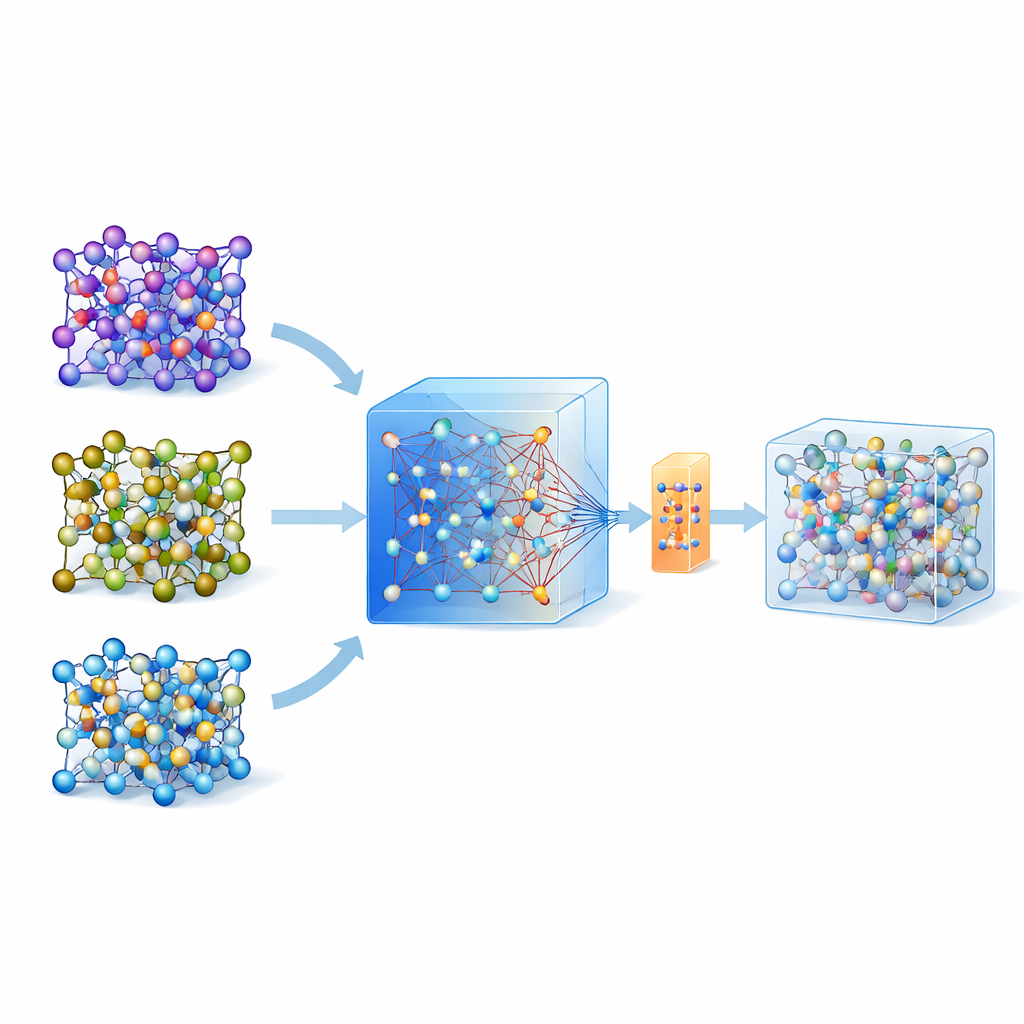
Perché simulare gli atomi è così difficile
Per valutare se un materiale solido trasporterà rapidamente gli ioni di litio, gli scienziati spesso ricorrono alla dinamica molecolare ab initio, una tecnica di riferimento che calcola il moto atomico dalla meccanica quantistica. Il problema è che è così dispendiosa dal punto di vista computazionale da non poter essere usata routinariamente per sistemi grandi o tempi lunghi. I potenziali interatomici basati su machine learning offrono una scorciatoia: una volta addestrati, imitano le forze quantistiche sottostanti a una frazione del costo. Tuttavia, costruire tali modelli per un materiale specifico ha tradizionalmente richiesto cicli complessi di “active learning” e migliaia o decine di migliaia di calcoli quantistici, limitandone fortemente la diffusione.
Usare un grande modello generale come guida
Negli ultimi anni sono emersi modelli universali e di grande scala addestrati su vasti database di calcoli quantistici su molti materiali. Uno di questi modelli, chiamato MACE-MP-0, è il punto di partenza qui. Gli autori hanno prima testato questo modello universale su tre elettroliti a stato solido di interesse tecnologico che coprono chimiche diverse: un solfuro (LGPS), un ossido (LATP) e un alogenuro (Li3YCl6). Hanno constatato che, mentre MACE-MP-0 poteva riprodurre grossolanamente le traiettorie atomiche delle costose simulazioni di riferimento, non prevedeva con sufficiente accuratezza proprietà delicate come le barriere alla migrazione del litio e i tassi di diffusione. Tuttavia, il suo esplorare lo spazio delle configurazioni atomiche corrispondeva strettamente ai calcoli di alto livello, rendendolo un eccellente e economico “campionatore” di strutture atomiche rilevanti.
Costruire modelli accurati da set di dati minimi
Invece di aggiornare ripetutamente un modello con molti cicli di active learning, gli autori propongono una strategia in singola fase. Prima eseguono dinamiche molecolari ad alta temperatura usando il modello universale MACE per generare molte istantanee atomiche. Poi applicano un metodo di riallocazione intelligente per scegliere solo circa 200 configurazioni particolarmente informative e calcolarne energie e forze con metodi quantistici completi. Piuttosto che addestrare un modello nuovo da zero, perfezionano il modello MACE esistente su questo set di dati piccolo ma selezionato con cura, usando sia aggiornamenti convenzionali sia una variante a efficienza parametrica chiamata ELoRA. Questo modello messo a punto non solo diventa significativamente più accurato per barriere energetiche e diffusione, ma eredita anche la stabilità dinamica del grande modello originale, evitando collassi atomici non fisici che spesso affliggono modelli addestrati con dati molto limitati.
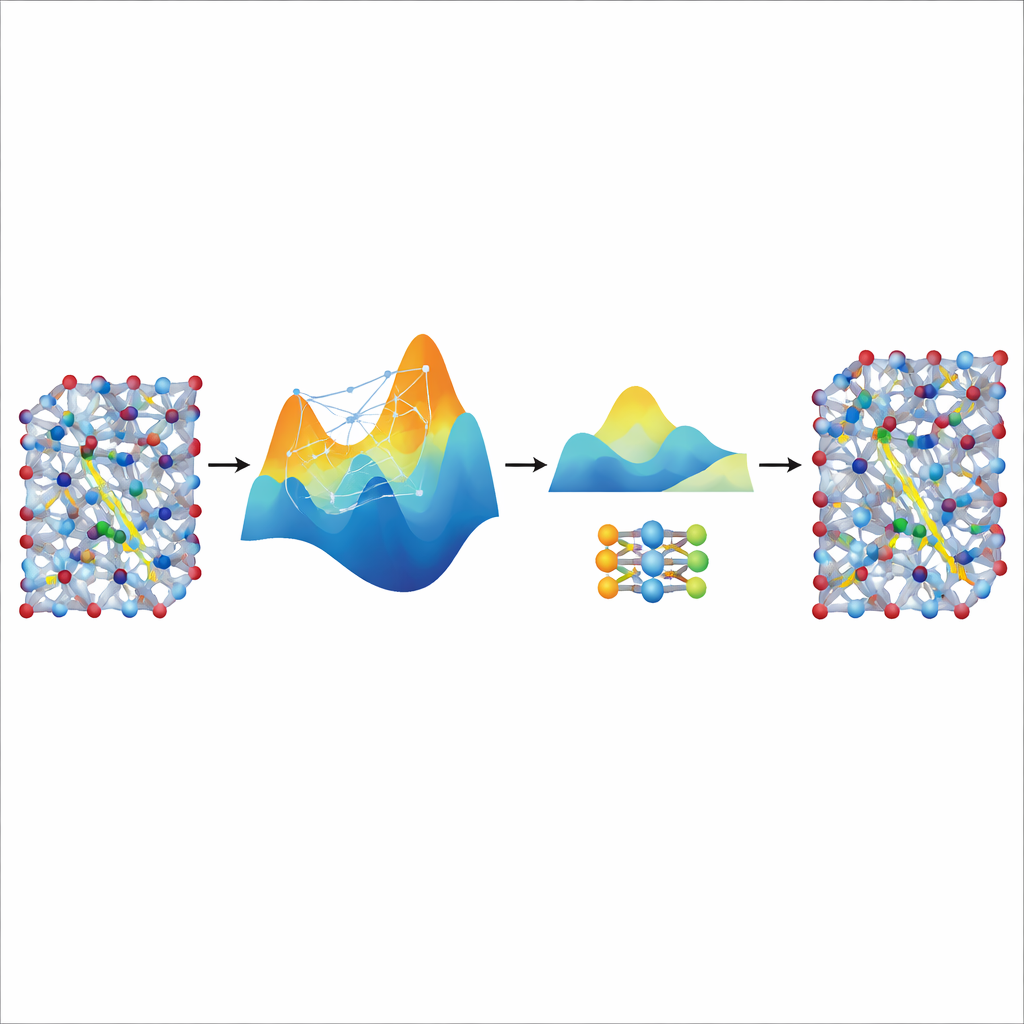
Distillare velocità da un grande insegnante
Sebbene il modello MACE messo a punto sia accurato e stabile, rimane relativamente pesante e lento per le simulazioni veramente lunghe e su larga scala necessarie a studiare il trasporto ionico in materiali per batterie realistici. Per risolvere questo, gli autori lo usano come “insegnante” per un modello molto più piccolo e leggero noto come NEP. Lasciando che il modello MACE perfezionato generi ulteriori dati di addestramento sintetici—migliaia di configurazioni atomiche etichettate con le sue energie e forze predette—senza altri calcoli quantistici, l’addestramento di NEP su questo dataset distillato produce un modello compatto che gira circa venti volte più veloce pur riproducendo strettamente le predizioni dell’insegnante. In grandi simulazioni con celle super, il modello NEP distillato riproduce caratteristiche chiave come transizioni superioniche e conduttività a temperatura ambiente in buon accordo con gli esperimenti.
Cosa significa questo per i materiali futuri
Lo studio dimostra una ricetta pratica per costruire campi di forza di machine learning affidabili e veloci usando solo poche centinaia di calcoli quantistici costosi: campionare ampiamente con un modello universale, affinarlo con cura e poi distillarne la conoscenza in uno studente più snello. Per gli elettroliti a stato solido, questo approccio consente simulazioni lunghe e su larga scala che catturano direttamente come gli ioni di litio si muovono attraverso strutture cristalline complesse, fornendo conduttività realistiche invece di stime grossolane. Più in generale, lo stesso flusso di lavoro potrebbe accelerare la progettazione di molti materiali funzionali, avvicinando il sogno di simulazioni atomistiche di alta fedeltà alla pratica quotidiana della ricerca.
Citazione: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Parole chiave: elettroliti a stato solido, potenziali di machine learning, dynamica molecolare, materiali per batterie, simulazione dei materiali