Clear Sky Science · sv
Konstruera maskininlärningsbaserade interatomära potentialer med minsta möjliga mängd ab initio-data
Smartare simuleringar för bättre batterier
Solid-state-batterier lovar säkrare mobiltelefoner, bilar och nätlagring genom att ersätta lättantändliga flytande elektrolyter med fasta material som leder litiumjoner. Men att hitta och testa nya fasta ledare är långsamt och kostsamt, särskilt när forskare förlitar sig på tunga superdatorkalkyler som följer varje elektron. Denna artikel visar hur modern maskininlärning kan dramatiskt minska den kostnaden: författarna bygger exakta, snabba ”digitala tvillingar” av atomära krafter med bara några hundra dyra beräkningar istället för tiotusentals, vilket öppnar för snabb screening av nästa generations batterimaterial.
Varför det är så svårt att simulera atomer
För att bedöma om ett fast material snabbt kan transportera litiumjoner vänder sig forskare ofta till ab initio-molekylär dynamik, en guldkantad teknik som beräknar atomrörelser från kvantmekanik. Problemet är att den är så beräkningskrävande att den inte kan användas rutinmässigt för stora system eller långa tider. Maskininlärningsbaserade interatomära potentialer erbjuder en genväg: när de väl är tränade efterliknar de de underliggande kvantkrafterna till en bråkdel av kostnaden. Att bygga sådana modeller för ett specifikt material har dock traditionellt krävts intrikata ”aktivinlärnings”-loopar och tusentals till tiotusentals kvantberäkningar, vilket kraftigt begränsar hur utbredda de kan bli.
Använda en stor generell modell som vägvisare
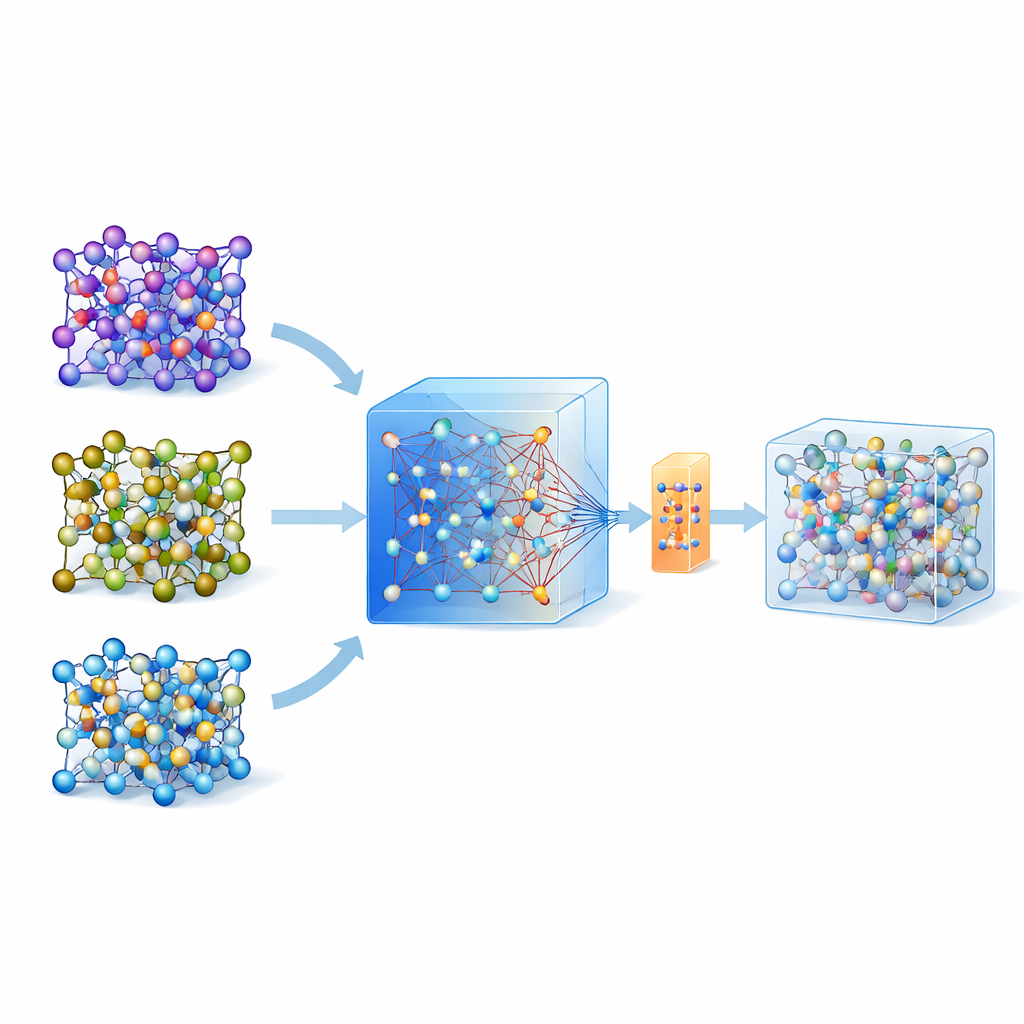
Under de senaste åren har universella, stora maskininlärningsmodeller tränade på enorma databaser av kvantberäkningar över många material fått genomslag. En sådan modell, kallad MACE-MP-0, fungerar här som utgångspunkt. Författarna testade först denna universella modell på tre tekniskt viktiga solid-state-elektrolyter som spänner över olika kemier: en sulfide (LGPS), en oxid (LATP) och en halid (Li3YCl6). De fann att även om MACE-MP-0 grovt kunde återge atombanorna från dyra referensimuleringar, förutsade den inte känsliga egenskaper som litiummigrationsbarriärer och diffusionshastigheter tillräckligt noggrant. Däremot matchade dess rörelser genom atomkonfigurationsrummet tätt med hög-nivå-beräkningarna, vilket gjorde den till en utmärkt, billig ”provtagare” av relevanta atomstrukturer.
Bygga exakta modeller från pyttesmå datamängder
I stället för att upprepade gånger uppdatera en modell med många rundor av aktiv inlärning föreslår författarna en engångsstrategi. Först kör de högtemperatur-molekylär dynamik med den universella MACE-modellen för att generera många atombilder. Sedan använder de en smart omsampling för att välja ut endast cirka 200 särskilt informativa konfigurationer och beräknar deras energier och krafter med fullständiga kvantmetoder. I stället för att träna en ny modell från grunden finjusterar de den befintliga MACE-modellen på denna lilla men omtänksamt utvalda datamängd, med både konventionell uppdatering och en parameter-effektiv variant kallad ELoRA. Denna finjusterade modell blir inte bara betydligt mer exakt för energibarriärer och diffusion, den ärv också den dynamiska stabiliteten hos den ursprungliga stora modellen, vilket undviker ofysisk atomkollaps som ofta plågar modeller tränade på mycket begränsade data.
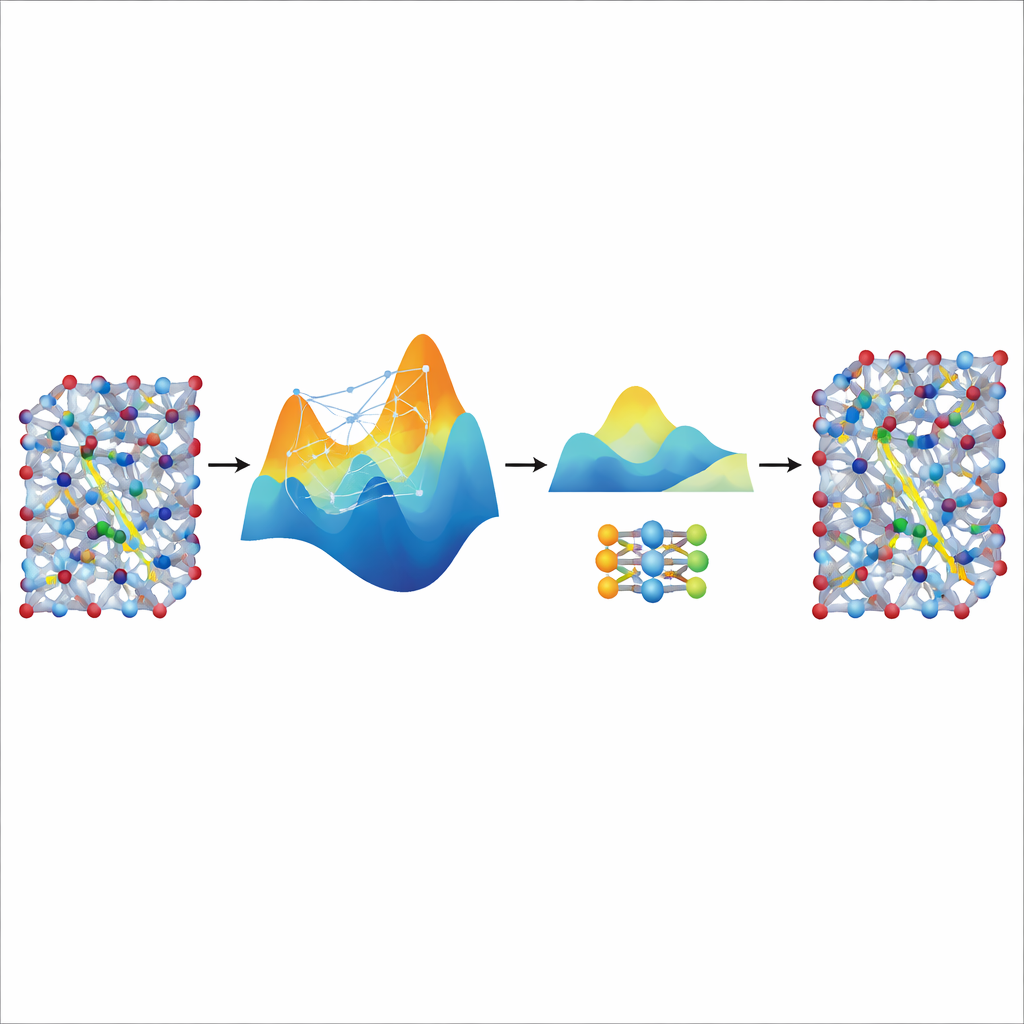
Destillera hastighet från en stor lärare
Även om den finjusterade MACE-modellen är exakt och stabil förblir den relativt tung och långsam för de verkligt långa och stora simuleringar som krävs för att studera jontransport i realistiska batterimaterial. För att lösa detta använder författarna den som en ”lärare” för en mycket mindre, lättviktsmodell känd som NEP. De låter den finjusterade MACE-modellen generera ytterligare syntetiska träningsdata—tusentals atomkonfigurationer märkta med dess förutsagda energier och krafter—utan några extra kvantberäkningar. Träning av NEP på denna destillerade datamängd ger en kompakt modell som kör ungefär tjugo gånger snabbare samtidigt som den nära matchar lärarens förutsägelser. I stora supercellsimuleringar återger den destillerade NEP-modellen nyckelfunktioner som superjoniska övergångar och konduktiviteter vid rumstemperatur som stämmer väl överens med experiment.
Vad detta betyder för framtida material
Studien visar ett praktiskt recept för att bygga tillförlitliga, snabba maskininlärningskraftfält med bara några hundra dyra kvantberäkningar: provta brett med en universell modell, finjustera den omsorgsfullt och destillera sedan dess kunskap till en slankare elev. För solid-state-elektrolyter möjliggör detta långa, storskaliga simuleringar som direkt fångar hur litiumjoner väver sig genom komplexa kristallstrukturer och ger realistiska konduktiviteter i stället för grova uppskattningar. Mer generellt kan samma arbetsflöde påskynda designen av många funktionella material och föra drömmen om rutinmässiga, högfidelitets atomistiska simuleringar mycket närmare vardaglig forskningspraxis.
Citering: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Nyckelord: solidstatelektrolyter, maskininlärningspotentials, molekylär dynamik, batterimaterial, materialssimulering