Clear Sky Science · pl
Budowa międzyatomowych potencjałów uczących się maszynowo przy minimalnej liczbie danych ab initio
Sprytniejsze symulacje dla lepszych baterii
Baterie w stanie stałym obiecują bezpieczniejsze telefony, samochody i magazyny energii sieciowej, zastępując łatwopalne elektrolity ciekłe stałymi materiałami przewodzącymi jony litu. Jednak odnajdywanie i testowanie nowych przewodników stałych jest powolne i kosztowne, zwłaszcza gdy badacze polegają na dużych obliczeniach superkomputerowych śledzących każdy elektron. Ten artykuł pokazuje, jak zastosować nowoczesne metody uczenia maszynowego, aby radykalnie obniżyć te koszty: autorzy budują dokładne, szybkie „cyfrowe bliźniaki” sił międzyatomowych przy użyciu zaledwie kilkuset drogich obliczeń zamiast dziesiątek tysięcy, otwierając drogę do szybkiego przesiewu materiałów nowej generacji do baterii.
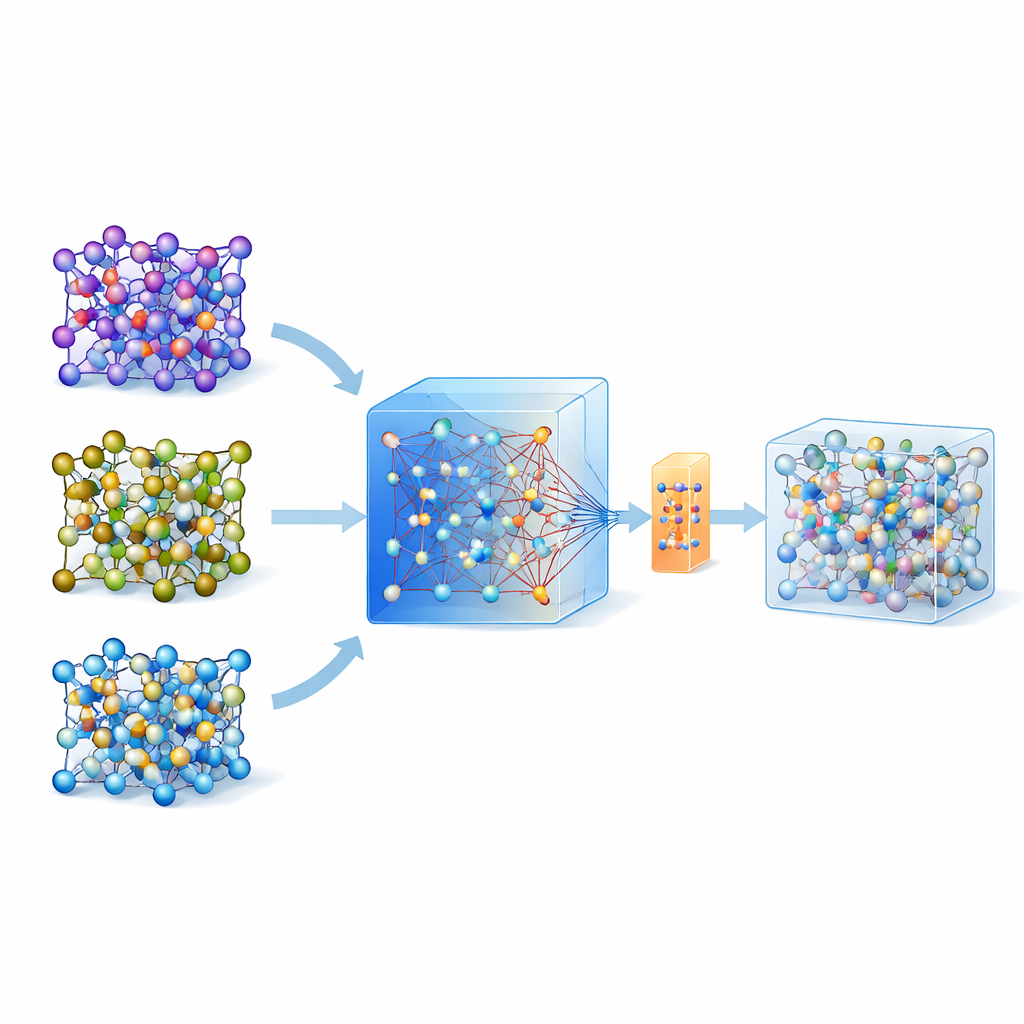
Dlaczego symulowanie atomów jest takie trudne
Aby ocenić, czy materiał stały będzie przewodził jony litu szybko, naukowcy często sięgają po dynamikę molekularną ab initio, technikę wzorcową, która oblicza ruch atomów na podstawie mechaniki kwantowej. Problem w tym, że jest ona tak wymagająca obliczeniowo, że nie może być rutynowo stosowana do dużych układów ani na długich czasach. Międzyatomowe potencjały oparte na uczeniu maszynowym oferują skrót: raz wytrenowane, naśladują siły kwantowe przy ułamku kosztu. Jednak tworzenie takich modeli dla konkretnego materiału tradycyjnie wymagało skomplikowanych pętli „aktywnego uczenia” i tysięcy do dziesiątek tysięcy obliczeń kwantowych, co znacznie ograniczało ich szersze zastosowanie.
Wykorzystanie dużego, ogólnego modelu jako przewodnika
W ostatnich latach pojawiły się uniwersalne, duże modele uczenia maszynowego trenowane na ogromnych bazach danych obliczeń kwantowych dla wielu materiałów. Jednym z takich modeli jest MACE-MP-0, który posłużył tu jako punkt wyjścia. Autorzy najpierw przetestowali ten uniwersalny model na trzech technologicznie istotnych elektrolitach w stanie stałym o różnych chemiach: na siarczku (LGPS), tlenku (LATP) i halidzie (Li3YCl6). Stwierdzili, że choć MACE-MP-0 potrafił w przybliżeniu odtworzyć trajektorie atomowe z drogich symulacji referencyjnych, nie przewidywał wystarczająco dokładnie subtelnych wielkości, takich jak bariery migracji litu i szybkości dyfuzji. Mimo to jego eksploracja przestrzeni konfiguracji atomowych była zbliżona do obliczeń wysokiego poziomu, co czyniło go doskonałym, tanim „próbkowaczem” istotnych struktur atomowych.
Budowanie dokładnych modeli z małych zestawów danych
Zamiast wielokrotnego aktualizowania modelu w wielu rundach aktywnego uczenia, autorzy proponują strategię jednokrotnego podejścia. Najpierw uruchamiają dynamikę molekularną w wysokiej temperaturze z użyciem uniwersalnego modelu MACE, aby wygenerować wiele migawkowych konfiguracji atomowych. Następnie stosują inteligentną metodę ponownego próbkowania, aby wybrać tylko około 200 szczególnie informatywnych konfiguracji i obliczyć ich energie oraz siły za pomocą pełnych metod kwantowych. Zamiast trenować nowy model od zera, dostrajają istniejący model MACE na tym małym, lecz starannie dobranym zbiorze danych, wykorzystując tradycyjne aktualizacje oraz efektywną parametrycznie odmianę zwaną ELoRA. Ten dostrojony model nie tylko staje się znacząco dokładniejszy dla barier energetycznych i dyfuzji, ale także dziedziczy stabilność dynamiczną oryginalnego dużego modelu, unikając niefizycznych kolapsów atomowych, które często dotykają modele trenowane na bardzo ograniczonych danych.
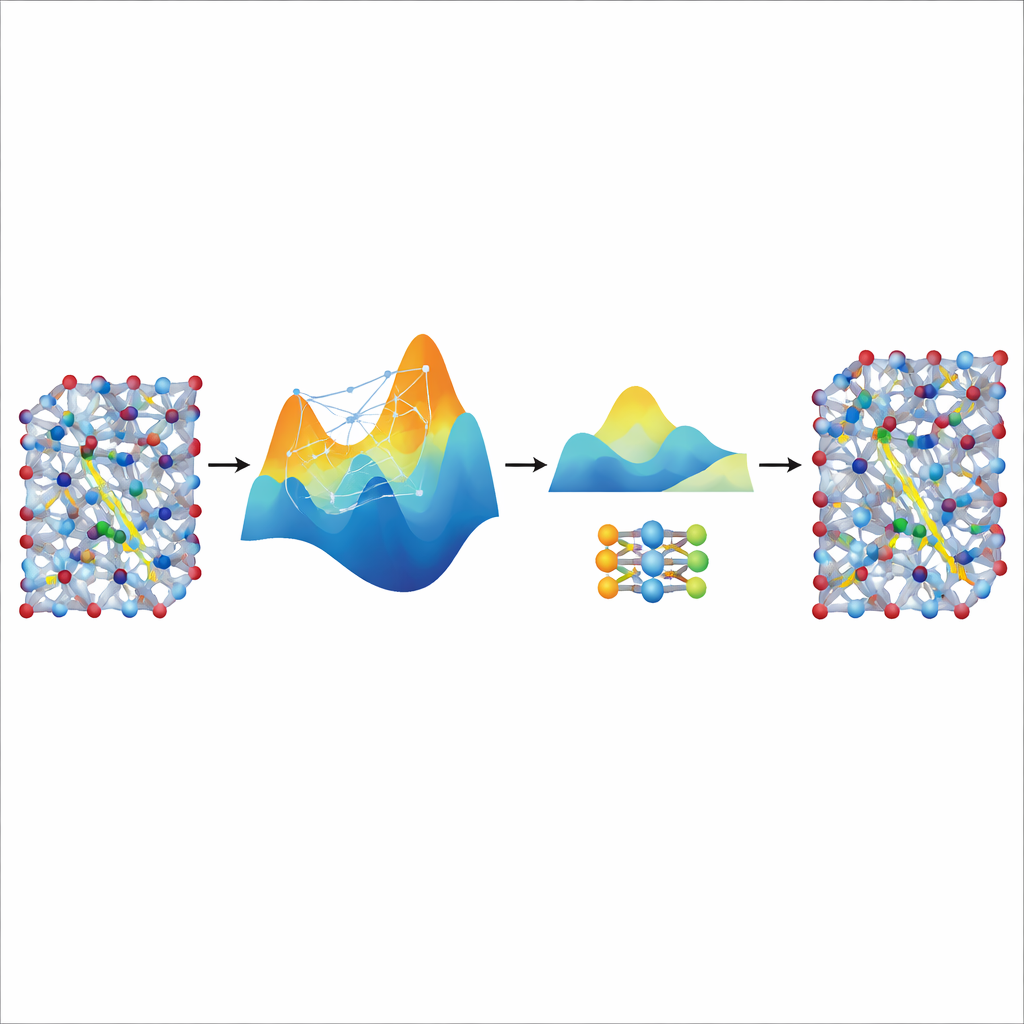
Destylacja szybkości od dużego nauczyciela
Choć dostrojony model MACE jest dokładny i stabilny, pozostaje stosunkowo ciężki i wolny do naprawdę długich i dużych symulacji potrzebnych do badania transportu jonów w realistycznych materiałach baterii. Aby temu zaradzić, autorzy używają go jako „nauczyciela” dla znacznie mniejszego, lekkiego modelu nazwanego NEP. Pozwalają dostrojonemu modelowi MACE wygenerować dodatkowe syntetyczne dane treningowe — tysiące konfiguracji atomowych oznaczonych jego przewidywanymi energiami i siłami — bez żadnych dodatkowych obliczeń kwantowych. Trenowanie NEP na tym zdestylowanym zbiorze danych daje kompaktowy model, który działa około dwadzieścia razy szybciej, zachowując jednocześnie zgodność z przewidywaniami nauczyciela. W dużych symulacjach superkomórkowych zdestylowany model NEP odtwarza kluczowe cechy, takie jak przejścia superjonowe i przewodności w temperaturze pokojowej, które dobrze zgadzają się z eksperymentami.
Co to oznacza dla przyszłych materiałów
Praca demonstruje praktyczny przepis na budowę niezawodnych, szybkich pól sił uczonych maszynowo przy użyciu zaledwie kilkuset drogich obliczeń kwantowych: szeroko próbkuj za pomocą uniwersalnego modelu, ostrożnie go dostrój, a następnie zdestyluj jego wiedzę do lżejszego modelu ucznia. Dla elektrolitów w stanie stałym takie podejście umożliwia długie, wielkoskalowe symulacje, które bezpośrednio pokazują, jak jony litu przemieszczają się przez złożone struktury krystaliczne, dostarczając realistycznych przewodności zamiast uproszczonych estymat. Szerzej, ten sam przepływ pracy może przyspieszyć projektowanie wielu funkcjonalnych materiałów, przybliżając marzenie o rutynowych, wysokiej wierności symulacjach atomistycznych do codziennej praktyki badawczej.
Cytowanie: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Słowa kluczowe: elektrolity w stanie stałym, potencjały uczone maszynowo, dynamika molekularna, materiały do baterii, symulacja materiałów