Clear Sky Science · ru
Построение межатомных потенциалов машинного обучения с минимальным количеством аб initio-данных
Умные симуляции для лучших батарей
Твердотельные батареи обещают более безопасные телефоны, автомобили и накопители для сети, заменив воспламеняющиеся жидкие электролиты твердыми материалами, проводящими ионы лития. Но поиск и проверка новых твердых проводников идут медленно и дорого, особенно когда исследователи полагаются на тяжёлые суперкомпьютерные расчёты, отслеживающие каждую электронную степень свободы. В этой работе показывают, как с помощью современных методов машинного обучения существенно сократить эти затраты: авторы создают точные, быстрые «цифровые двойники» атомных сил, используя всего несколько сотен дорогих расчётов вместо десятков тысяч, что открывает путь к быстрому скринингу материалов следующего поколения для батарей.
Почему моделировать атомы так сложно
Чтобы оценить, будет ли твердый материал быстро переносить ионы лития, учёные часто обращаются к ab initio молекулярной динамике — эталонной технике, вычисляющей движение атомов из законов квантовой механики. Проблема в том, что она настолько требовательна к вычислительным ресурсам, что её нельзя регулярно применять к большим системам или на длинных временных шкалах. Межатомные потенциалы на основе машинного обучения предлагают обходной путь: однажды обученные, они воспроизводят квантовые силы с долей расходов. Однако построение таких моделей для конкретного материала традиционно требовало сложных циклов «активного обучения» и тысяч — десятков тысяч квантовых расчётов, что сильно ограничивало их широкое применение.
Использование большой общей модели как ориентира
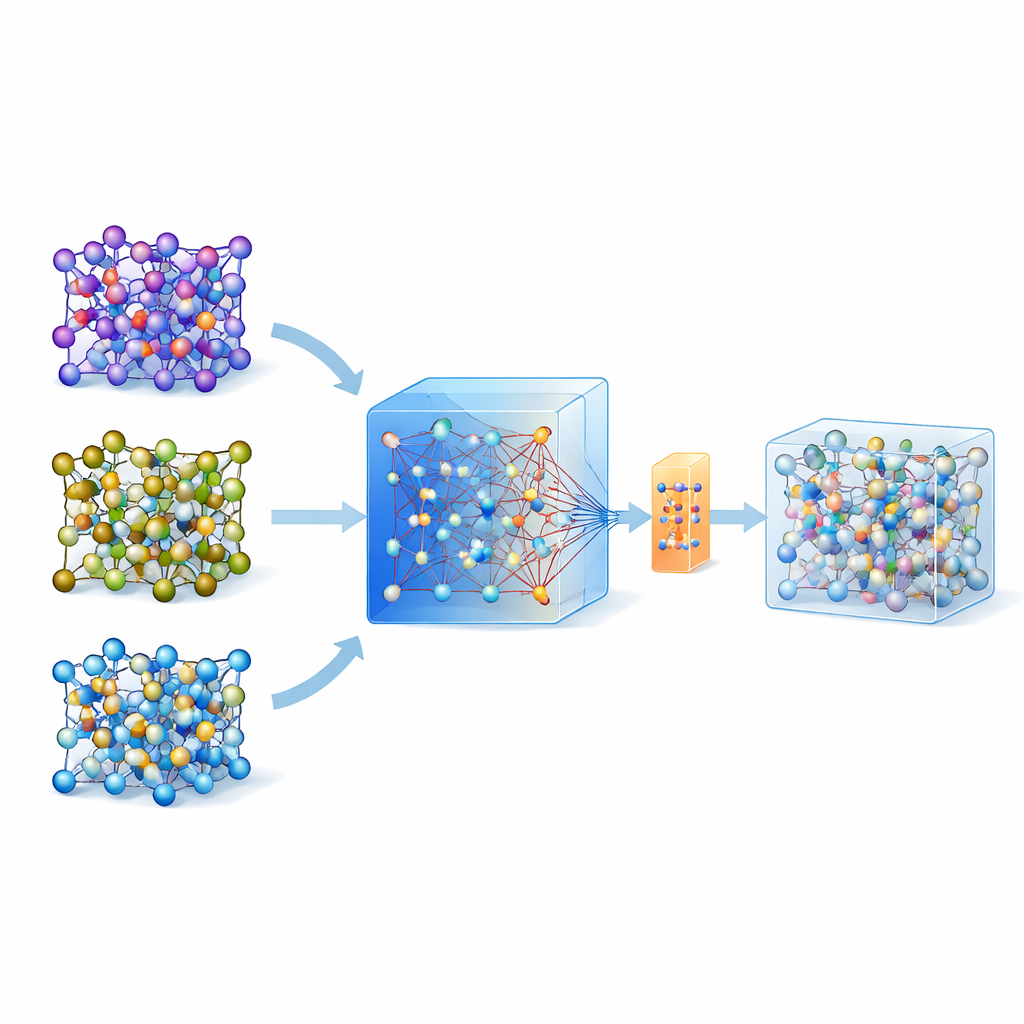
В последние годы появились универсальные крупные модели машинного обучения, обученные на огромных базах квантовых расчётов по множеству материалов. Одна из таких моделей, называемая MACE-MP-0, используется здесь в качестве отправной точки. Авторы сначала протестировали эту универсальную модель на трёх технологически важных твердотельных электролитах с разной химией: сульфиде (LGPS), оксиде (LATP) и галогениде (Li3YCl6). Они обнаружили, что хотя MACE-MP-0 в целом может приближённо воспроизводить атомные траектории из дорогих эталонных симуляций, она недостаточно точно предсказывает тонкие свойства, такие как барьеры миграции лития и коэффициенты диффузии. Тем не менее её движение по пространству атомных конфигураций тесно соответствовало высокоуровневым расчётам, делая её отличным и недорогим «сэмплером» релевантных атомных структур.
Построение точных моделей из крошечных наборов данных
Вместо многократного обновления модели в циклах активного обучения авторы предлагают стратегию «одного выстрела». Сначала они проводят молекулярную динамику при высокой температуре, используя универсальную модель MACE, чтобы сгенерировать множество атомных снимков. Затем применяют умный метод повторной выборки, чтобы отобрать примерно 200 особенно информативных конфигураций и вычислить их энергии и силы полными квантовыми методами. Вместо обучения новой модели с нуля они дообучают существующую модель MACE на этом небольшом, но тщательно отобранном наборе данных, применяя как обычное обновление параметров, так и параметро-экономичную версию, называемую ELoRA. Такая донастройка не только значительно повышает точность предсказаний барьеров и диффузии, но и сохраняет динамическую стабильность исходной крупной модели, избегая нефизических «коллапсов» атомных структур, которые часто возникают у моделей, обученных на очень ограниченных данных.
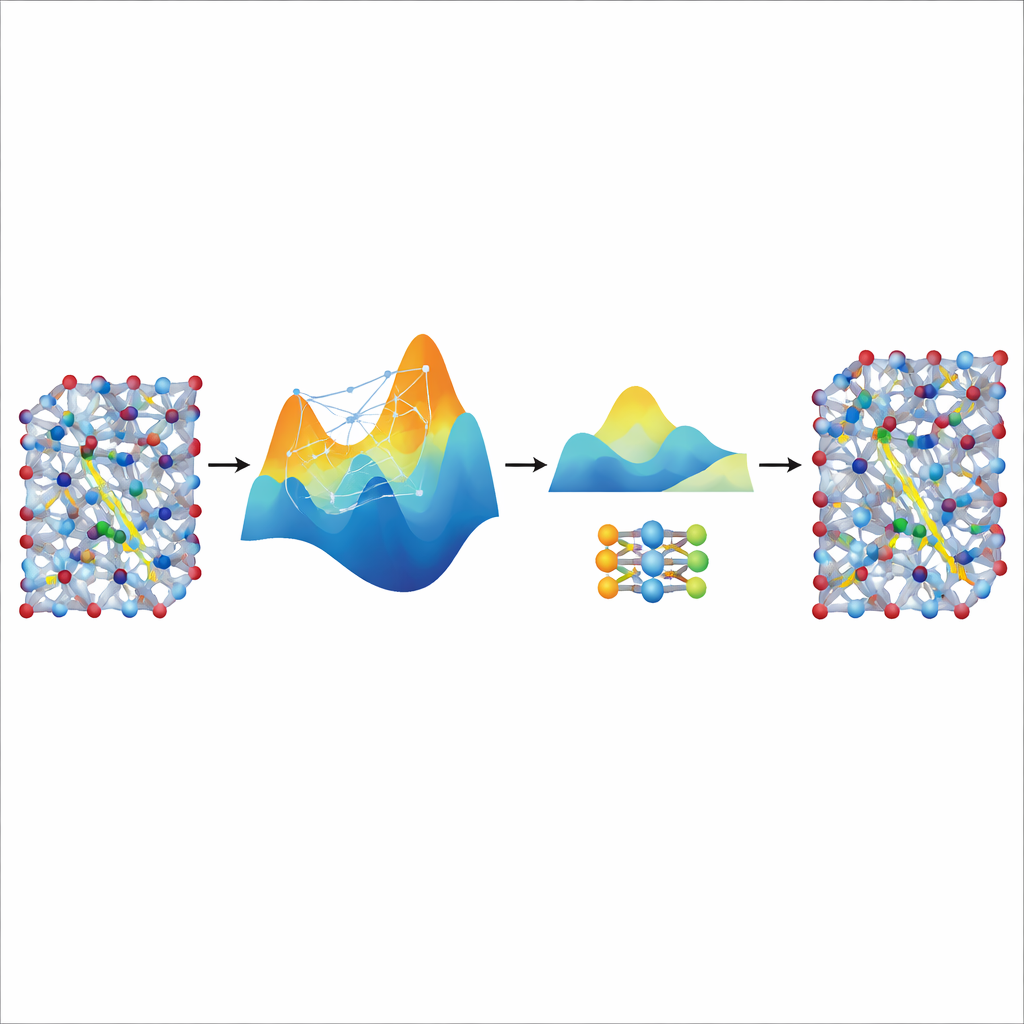
Перенос скорости от большого учителя
Хотя донастроенная модель MACE точна и стабильна, она всё ещё остаётся относительно тяжёлой и медленной для действительно длинных и крупных симуляций, необходимых для изучения транспорта ионов в реалистичных материалах для батарей. Чтобы решить эту проблему, авторы используют её как «учителя» для гораздо меньшей, лёгкой модели, известной как NEP. Они позволяют настроенной модели MACE сгенерировать дополнительный синтетический тренировочный набор — тысячи атомных конфигураций с пометками её предсказанных энергий и сил — без каких-либо дополнительных квантовых расчётов. Обучение NEP на этом дистиллированном наборе данных даёт компактную модель, работающую примерно в двадцать раз быстрее при тесном соответствии предсказаниям учителя. В крупных суперячейковых симуляциях дистиллированная модель NEP воспроизводит ключевые особенности, такие как суперионные переходы и проводимости при комнатной температуре, хорошо согласующиеся с экспериментами.
Что это значит для материалов будущего
Исследование демонстрирует практичный рецепт создания надёжных и быстрых полей сил машинного обучения, используя всего несколько сотен дорогих квантовых расчётов: широко сэмплировать с помощью универсальной модели, аккуратно её донастроить и затем дистиллировать знания в более лёгкую «студенческую» модель. Для твердотельных электролитов такой подход позволяет проводить длинные, крупномасштабные симуляции, которые напрямую фиксируют, как ионы лития протекают через сложные кристаллические структуры, давая реалистичные значения проводимости вместо грубых оценок. Более широко, тот же рабочий процесс может ускорить разработку многих функциональных материалов, приближая мечту о рутинном, высокоточном атомистическом моделировании к повседневной исследовательской практике.
Цитирование: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Ключевые слова: твердотельные электролиты, потенциалы машинного обучения, молекулярная динамика, материалы для батарей, моделирование материалов