Clear Sky Science · es
Construcción de potenciales interatómicos mediante aprendizaje automático con la mínima cantidad de datos ab initio
Simulaciones más inteligentes para mejores baterías
Las baterías en estado sólido prometen dispositivos más seguros —teléfonos, coches y almacenamiento en red— al reemplazar electrolitos líquidos inflamables por materiales sólidos que conducen iones de litio. Pero encontrar y probar nuevos conductores sólidos es lento y caro, sobre todo cuando los investigadores dependen de costosos cálculos en superordenadores que siguen cada electrón. Este artículo muestra cómo usar el aprendizaje automático moderno para reducir drásticamente ese coste: los autores construyen “gemelos digitales” rápidos y precisos de las fuerzas atómicas usando solo unos pocos cientos de cálculos costosos en lugar de decenas de miles, abriendo la puerta al cribado rápido de materiales de baterías de próxima generación.
Por qué es tan difícil simular átomos
Para evaluar si un material sólido transportará iones de litio con rapidez, los científicos suelen recurrir a la dinámica molecular ab initio, una técnica de referencia que calcula el movimiento atómico desde la mecánica cuántica. El inconveniente es que exige tanto cálculo que no puede emplearse de forma rutinaria en sistemas grandes o tiempos largos. Los potenciales interatómicos por aprendizaje automático ofrecen un atajo: una vez entrenados, imitan las fuerzas cuánticas subyacentes a una fracción del coste. Sin embargo, construir tales modelos para un material específico ha requerido tradicionalmente bucles intrincados de “aprendizaje activo” y miles o decenas de miles de cálculos cuánticos, lo que limita mucho su despliegue.
Usar un gran modelo general como guía
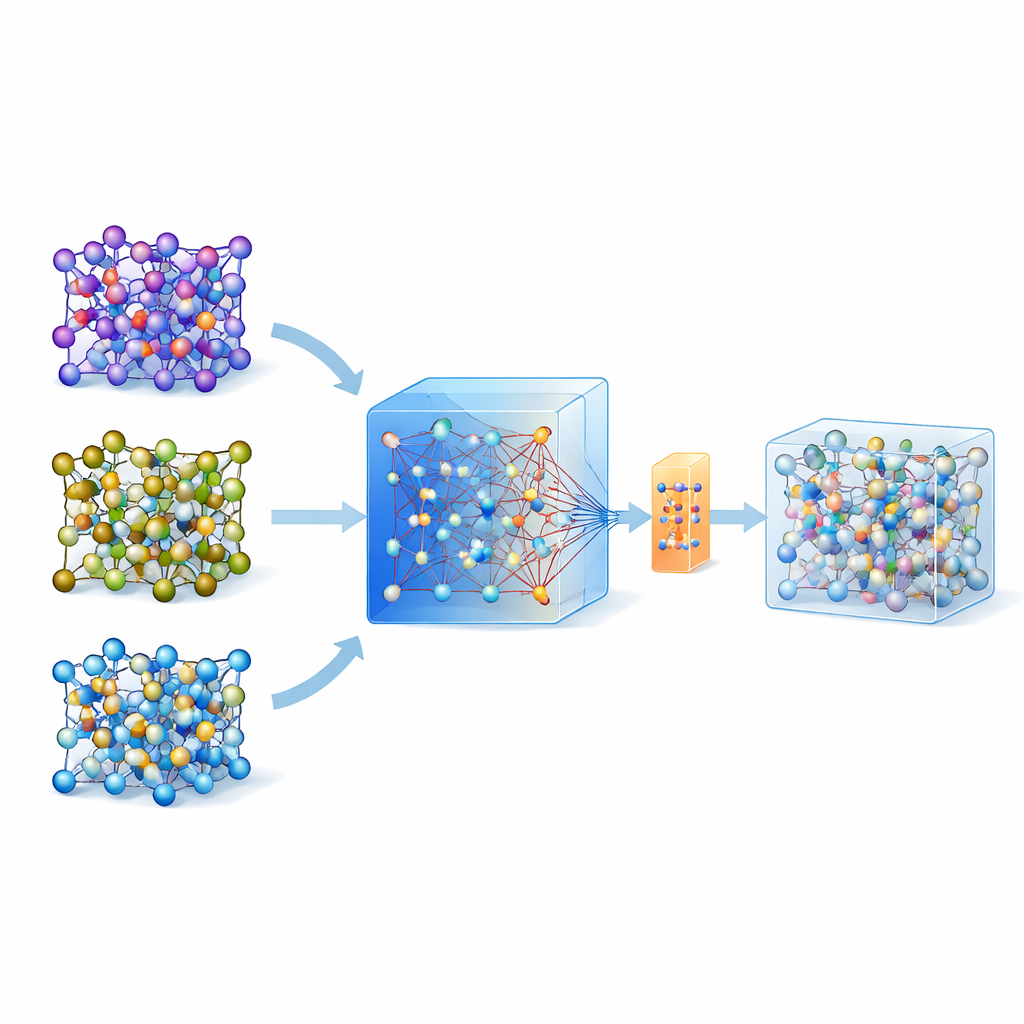
En los últimos años han emergido modelos universales y grandes entrenados en enormes bases de datos de cálculos cuánticos sobre muchos materiales. Uno de esos modelos, llamado MACE-MP-0, sirve aquí como punto de partida. Los autores probaron primero este modelo universal en tres electrolitos en estado sólido de interés tecnológico que cubren diferentes químicas: un sulfuro (LGPS), un óxido (LATP) y un haluro (Li3YCl6). Encontraron que, si bien MACE-MP-0 podía reproducir de forma aproximada las trayectorias atómicas de las simulaciones de referencia costosas, no predijo con suficiente precisión propiedades delicadas como las barreras de migración del litio y las tasas de difusión. Aun así, su recorrido por el espacio de configuraciones atómicas coincidía de forma estrecha con los cálculos de alto nivel, lo que lo convierte en un excelente y barato “muestreador” de estructuras atómicas relevantes.
Construir modelos precisos a partir de conjuntos de datos diminutos
En lugar de actualizar repetidamente un modelo con numerosas rondas de aprendizaje activo, los autores proponen una estrategia de una sola pasada. Primero ejecutan dinámica molecular a alta temperatura usando el modelo universal MACE para generar muchas instantáneas atómicas. Luego aplican un método inteligente de re-muestreo para elegir solo unas ~200 configuraciones especialmente informativas y calculan sus energías y fuerzas con métodos cuánticos completos. En lugar de entrenar un modelo nuevo desde cero, afinan el modelo MACE existente con este conjunto pequeño pero cuidadosamente seleccionado, utilizando tanto actualizaciones convencionales como una variante eficiente en parámetros llamada ELoRA. Este modelo ajustado no solo mejora de forma significativa la precisión en barreras energéticas y difusión, sino que también hereda la estabilidad dinámica del gran modelo original, evitando colapsos atómicos no físicos que suelen afectar a modelos entrenados con datos muy limitados.
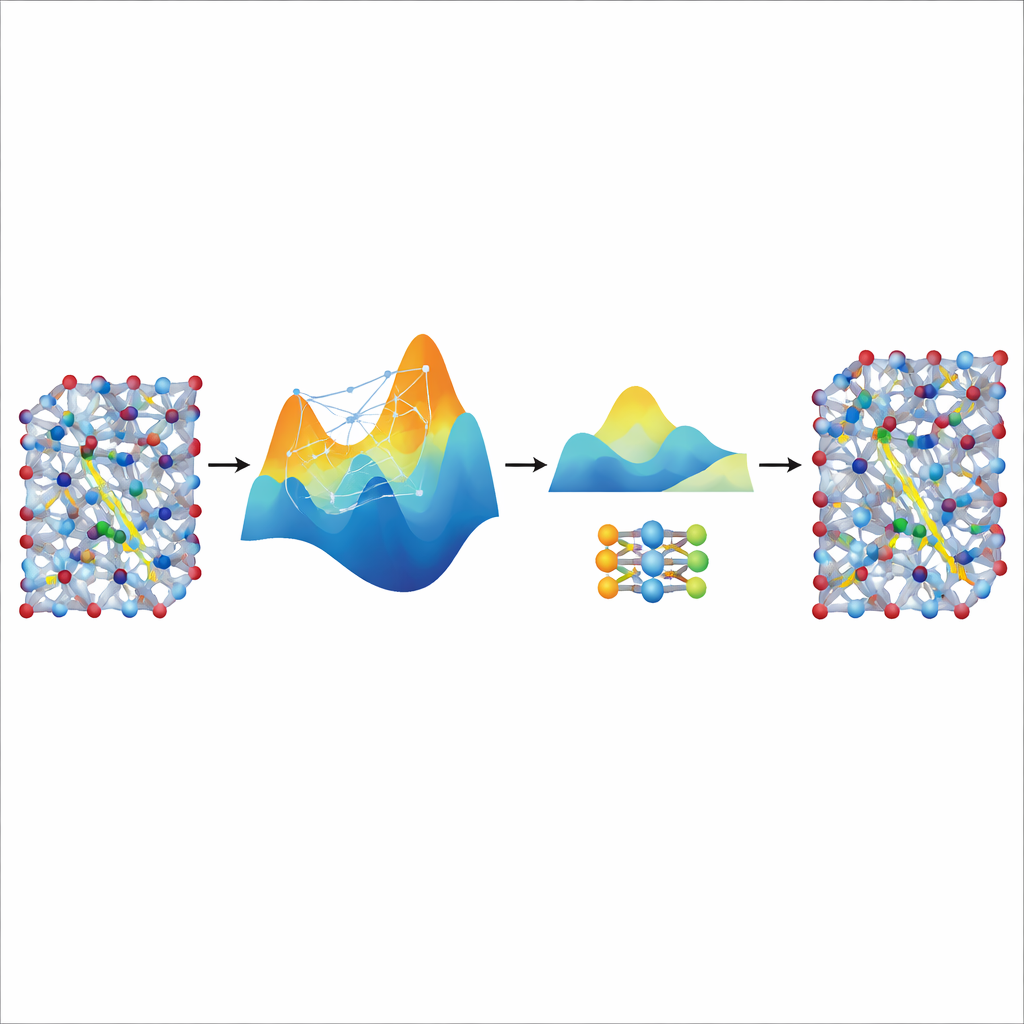
Destilar velocidad a partir de un gran maestro
Aunque el modelo MACE afinado es preciso y estable, sigue siendo relativamente pesado y lento para las simulaciones verdaderamente largas y grandes necesarias para estudiar el transporte iónico en materiales de baterías realistas. Para resolver esto, los autores lo emplean como “maestro” para un modelo mucho más pequeño y ligero conocido como NEP. Permiten que el MACE ajustado genere datos sintéticos de entrenamiento adicionales —miles de configuraciones atómicas etiquetadas con sus energías y fuerzas predichas— sin cálculos cuánticos adicionales. Entrenar NEP con este conjunto destilado produce un modelo compacto que funciona aproximadamente veinte veces más rápido, manteniendo un buen ajuste a las predicciones del maestro. En simulaciones con superceldas grandes, el modelo NEP destilado reproduce características clave como transiciones superiónicas y conductividades a temperatura ambiente que concuerdan bien con experimentos.
Qué significa esto para los materiales del futuro
El estudio demuestra una receta práctica para construir campos de fuerza por aprendizaje automático fiables y rápidos empleando solo unos pocos cientos de cálculos cuánticos costosos: muestrear ampliamente con un modelo universal, afinarlo con cuidado y luego destilar su conocimiento en un alumno más esbelto. Para los electrolitos en estado sólido, este enfoque permite simulaciones largas y a gran escala que capturan directamente cómo los iones de litio se abren paso a través de complejas estructuras cristalinas, proporcionando conductividades realistas en lugar de estimaciones burdas. Más ampliamente, el mismo flujo de trabajo podría acelerar el diseño de muchos materiales funcionales, acercando el sueño de la simulación atomística de alta fidelidad a la práctica investigadora cotidiana.
Cita: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Palabras clave: electrolitos en estado sólido, potenciales por aprendizaje automático, dinámica molecular, materiales para baterías, simulación de materiales