Clear Sky Science · ja
最小限の第一原理データで機械学習原子間ポテンシャルを構築する
より賢いシミュレーションでより良い電池を
固体電解質は可燃性の液体電解質をリチウムイオンを伝導する固体材料に置き換えることで、携帯電話や自動車、電力系統の蓄電をより安全にする可能性があります。しかし、新しい固体導体を見つけて評価する作業は、特に各電子を追跡する重いスーパーコンピュータ計算に依存する場合、遅く高価です。本論文は、現代の機械学習を用いることでそのコストを劇的に削減する方法を示します。著者らは、数万件ではなく数百件の高精度計算だけで原子力を正確かつ高速に再現する「デジタルツイン」を構築し、次世代電池材料の迅速なスクリーニングへの道を開きます。
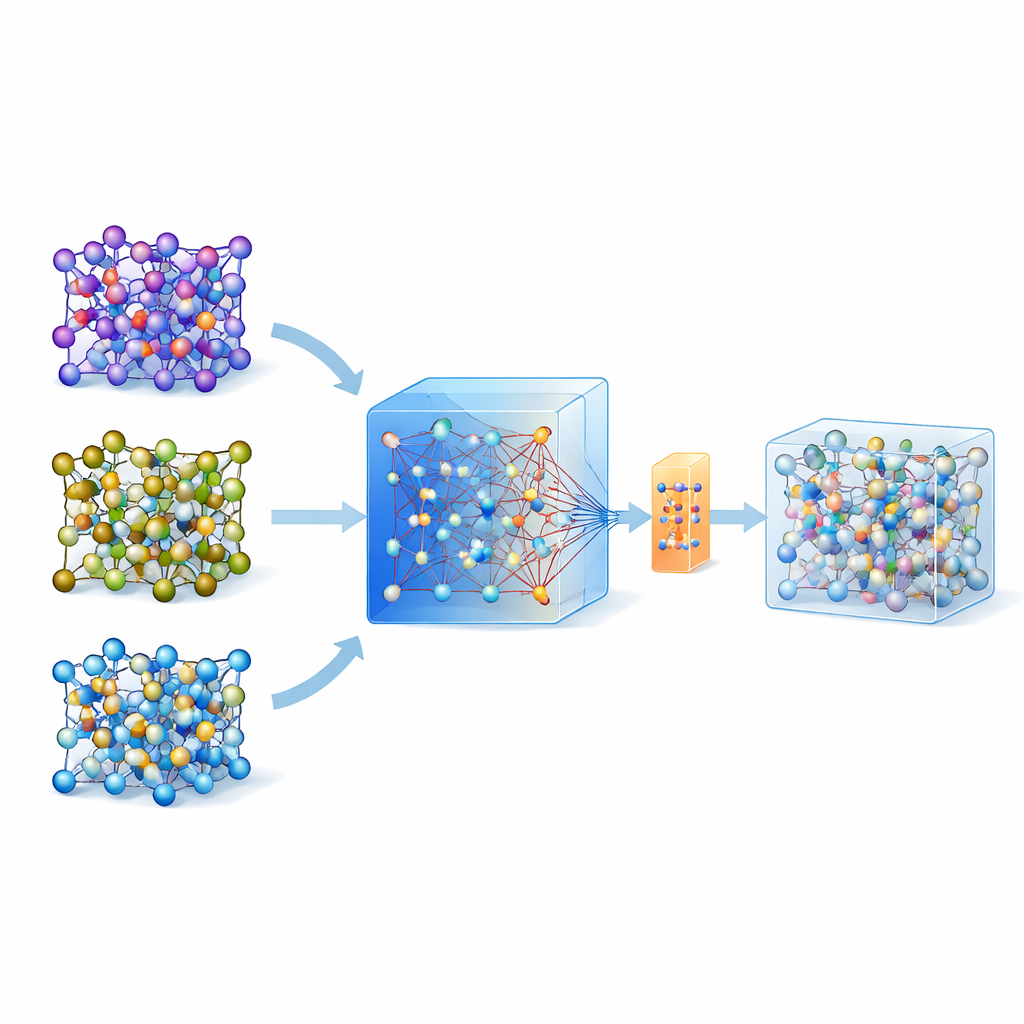
原子をシミュレートするのが難しい理由
固体材料がリチウムイオンを速やかに運ぶかを評価するために、研究者はしばしばab initio分子動力学に頼ります。これは原子運動を量子力学から計算するゴールドスタンダードの手法です。問題は、計算負荷が非常に大きく、大きな系や長時間のシミュレーションに日常的には使えない点です。機械学習原子間ポテンシャルは近道を提供します:一度学習させれば、基礎となる量子力学的な力をはるかに低コストで模倣できます。しかし、特定の材料向けにそのようなモデルを構築するには、従来は複雑なアクティブラーニングのループと何千〜何万という量子計算が必要であり、普及を大きく制限していました。
大きな汎用モデルをガイドとして使う
近年、多数の材料にわたる膨大な量子計算データベースで学習された、汎用で大規模な機械学習モデルが登場しています。本研究で出発点として用いられる一例が MACE-MP-0 というモデルです。著者らはまず、この汎用モデルを三種の技術的に重要な固体電解質(硫化物のLGPS、酸化物のLATP、ハライドのLi3YCl6)で試しました。MACE-MP-0は高精度の参照シミュレーションから得られる原子軌跡を概ね再現できましたが、リチウムの移動障壁や拡散率のような微妙な特性は十分に正確に予測できませんでした。それでも、原子配位空間を通るその運動は高レベル計算と密接に一致しており、関連する原子構造を安価にサンプリングする優れた手段となりました。
小さなデータセットから正確なモデルを作る
著者らは多数のアクティブラーニング反復を行う代わりに、ワンショット戦略を提案します。まず、汎用のMACEモデルを用いて高温分子動力学を走らせ、多数の原子スナップショットを生成します。次に、スマートな再サンプリング手法で特に情報量の多い約200の構成だけを選び、それらのエネルギーと力を完全な量子計算で評価します。新しいモデルを一から訓練するのではなく、既存のMACEモデルをこの小規模かつ慎重に選ばれたデータセットでファインチューニングします。通常の更新に加え、ELoRAと呼ばれるパラメータ効率の良い変種も用います。この調整済みモデルは、エネルギー障壁や拡散に関する精度が大幅に向上するだけでなく、元の大規模モデルが持つ動的安定性も受け継ぎ、非常に限られたデータで訓練した際に起こりがちな非現実的な原子崩壊を回避します。
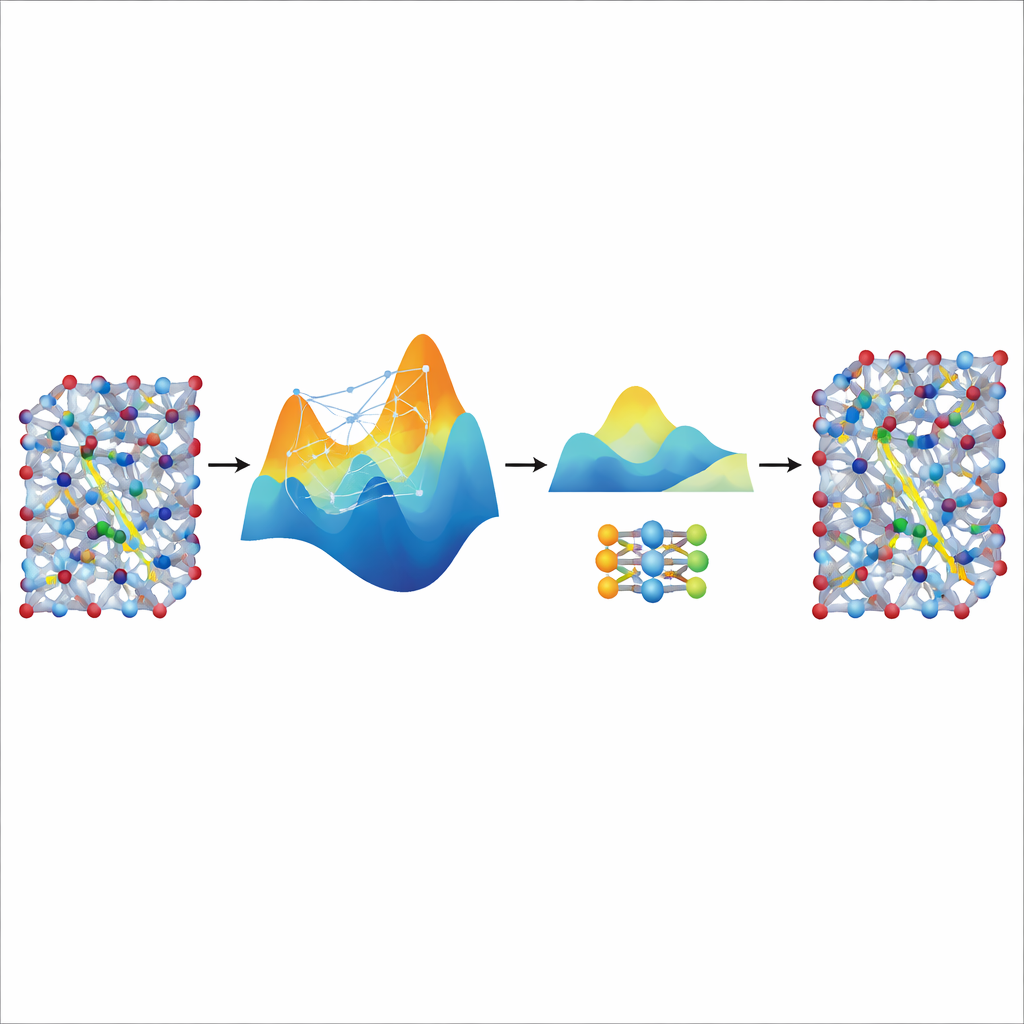
大きな教師から速度を蒸留する
ファインチューニングしたMACEモデルは精度と安定性を備えていますが、現実的な電池材料でのイオン輸送を研究するために必要な非常に長く大きなシミュレーションでは依然として重く遅いままです。そこで著者らはそれを「教師」として、NEPと呼ばれるはるかに小型で軽量なモデルに利用します。調整済みのMACEモデルに追加の合成トレーニングデータ—自身の予測するエネルギーと力でラベル付けされた何千もの原子構成—を生成させ、追加の量子計算を行うことなくデータを得ます。この蒸留されたデータでNEPを訓練すると、教師の予測に密接に一致しつつ約20倍速く動作するコンパクトなモデルが得られます。大きなスーパーセルシミュレーションでは、蒸留されたNEPモデルは超イオン伝導転移や室温伝導率といった実験と整合する主要な特徴を再現しました。
今後の材料研究にとっての意義
本研究は、数百件の高価な量子計算しか用いずに信頼できる高速な機械学習力場を構築する実用的なレシピを示します:汎用モデルで広くサンプリングし、丁寧にファインチューニングし、その知識をより軽量な生徒モデルに蒸留する。固体電解質に関しては、この手法によりリチウムイオンが複雑な結晶構造の中をどう巡るかを直接捉える長期・大規模シミュレーションが可能になり、粗い推定ではなく現実的な伝導率を提供します。より広く見れば、同様のワークフローは多くの機能性材料設計を加速し、高忠実度の原子スケールシミュレーションを日常の研究実践により近づけるでしょう。
引用: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
キーワード: 固体電解質, 機械学習ポテンシャル, 分子動力学, 電池材料, 材料シミュレーション