Clear Sky Science · nl
Opbouw van machine learning- tussenatomaire potentialen met minimale hoeveelheid ab initio-gegevens
Slimmere simulaties voor betere batterijen
Vaste-stofbatterijen beloven veiligere telefoons, auto’s en netopslag door brandbare vloeibare elektrolyten te vervangen door vaste materialen die lithiumionen geleiden. Maar het vinden en testen van nieuwe vaste geleiders is traag en duur, vooral wanneer onderzoekers vertrouwen op zware supercomputerberekeningen die elke elektron volgen. Dit artikel laat zien hoe moderne machine learning die kosten drastisch kan verlagen: de auteurs bouwen nauwkeurige, snelle “digitale tweelingen” van atomaire krachten met slechts enkele honderden dure berekeningen in plaats van tienduizenden, waardoor snelle screening van de volgende generatie batterijmaterialen mogelijk wordt.
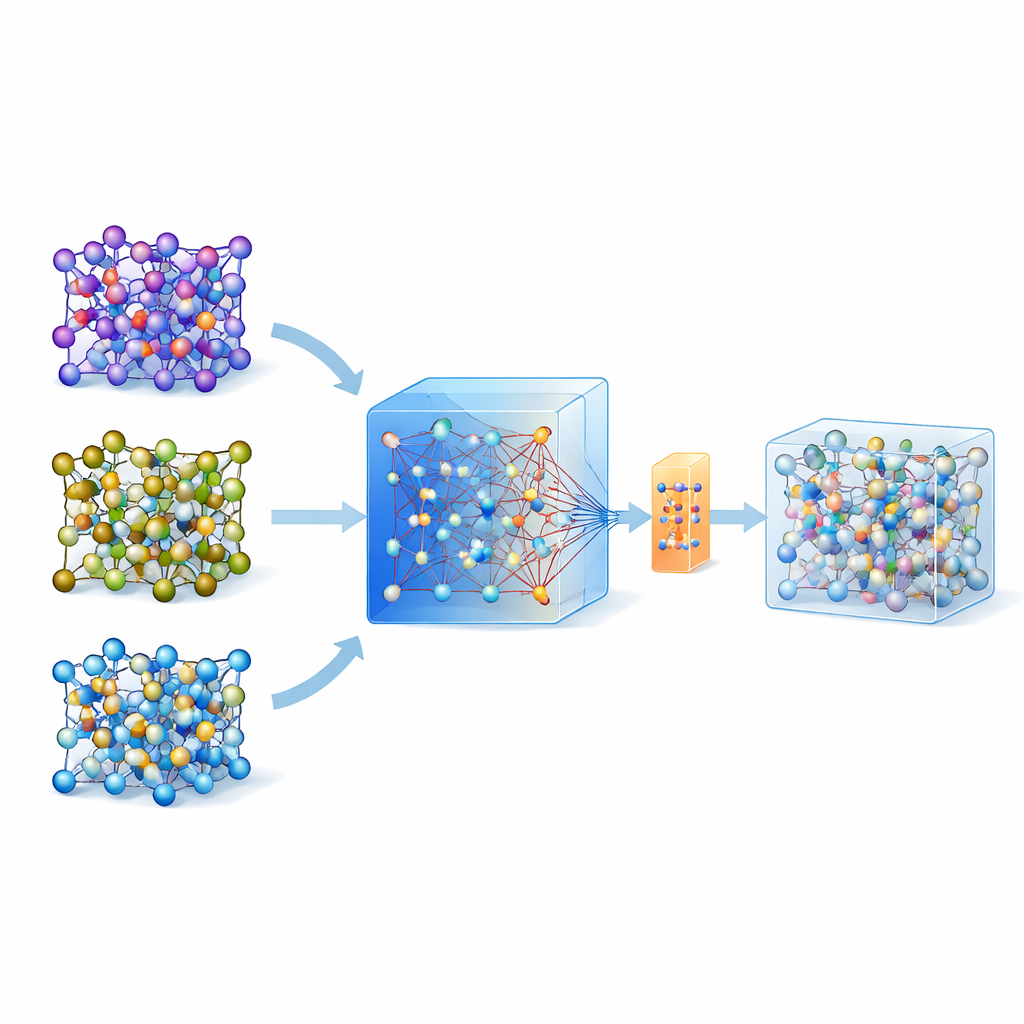
Waarom atomair simuleren zo moeilijk is
Om te beoordelen of een vast materiaal lithiumionen snel zal transporteren, grijpen wetenschappers vaak naar ab initio moleculaire dynamica, een goudstandaardtechniek die atomaire beweging uit de kwantummechanica berekent. Het probleem is dat dit zo rekenintensief is dat het niet routinematig toepasbaar is op grote systemen of lange tijden. Machine learning tussenatomaire potentialen bieden een omweg: eenmaal getraind imiteren ze de onderliggende kwantumkrachten tegen een fractie van de kosten. Het opbouwen van zulke modellen voor een specifiek materiaal vereiste traditioneel echter ingewikkelde “active learning”-lussen en duizenden tot tienduizenden kwantumberekeningen, wat hun brede inzet sterk beperkt.
Een groot algemeen model als gids gebruiken
De laatste jaren zijn universele, grote machine-learningmodellen opgekomen die zijn getraind op enorme databases van kwantumberekeningen voor veel materialen. Eén zo’n model, genoemd MACE-MP-0, dient hier als uitgangspunt. De auteurs testten dit universele model eerst op drie technologisch belangrijke vaste-stof elektrolyten die verschillende chemieën beslaan: een sulfide (LGPS), een oxide (LATP) en een halide (Li3YCl6). Ze ontdekten dat hoewel MACE-MP-0 ruwweg de atomaire trajecten van de dure referentiesimulaties kon reproduceren, het delicate eigenschappen zoals lithiummigratiebarrières en diffusiesnelheden niet nauwkeurig genoeg voorspelde. Toch kwam zijn beweging door de configuratieruimte van atomen sterk overeen met die van de hoogwaardige berekeningen, waardoor het een uitstekende en goedkope “sampler” van relevante atomaire structuren is.
Nauwkeurige modellen bouwen uit kleine datasets
In plaats van een model herhaaldelijk bij te werken met vele rondes van active learning, stellen de auteurs een single-shot strategie voor. Eerst voeren ze hogetemperatuursmoleculaire dynamica uit met het universele MACE-model om veel atomaire snapshots te genereren. Daarna passen ze een slimme resampleermethode toe om slechts ongeveer 200 bijzonder informatieve configuraties te selecteren en berekenen hun energieën en krachten met volledige kwantummethoden. In plaats van een nieuw model vanaf nul te trainen, finetunen ze het bestaande MACE-model op deze kleine maar zorgvuldig gekozen dataset, gebruikmakend van zowel conventionele updates als een parameterefficiënte variant genaamd ELoRA. Dit afgestemde model wordt niet alleen aanzienlijk nauwkeuriger voor energiebarrières en diffusie, het erft ook de dynamische stabiliteit van het oorspronkelijke grote model, waardoor onfysische atomaire instortingen die modellen met zeer beperkte data vaak teisteren, worden vermeden.
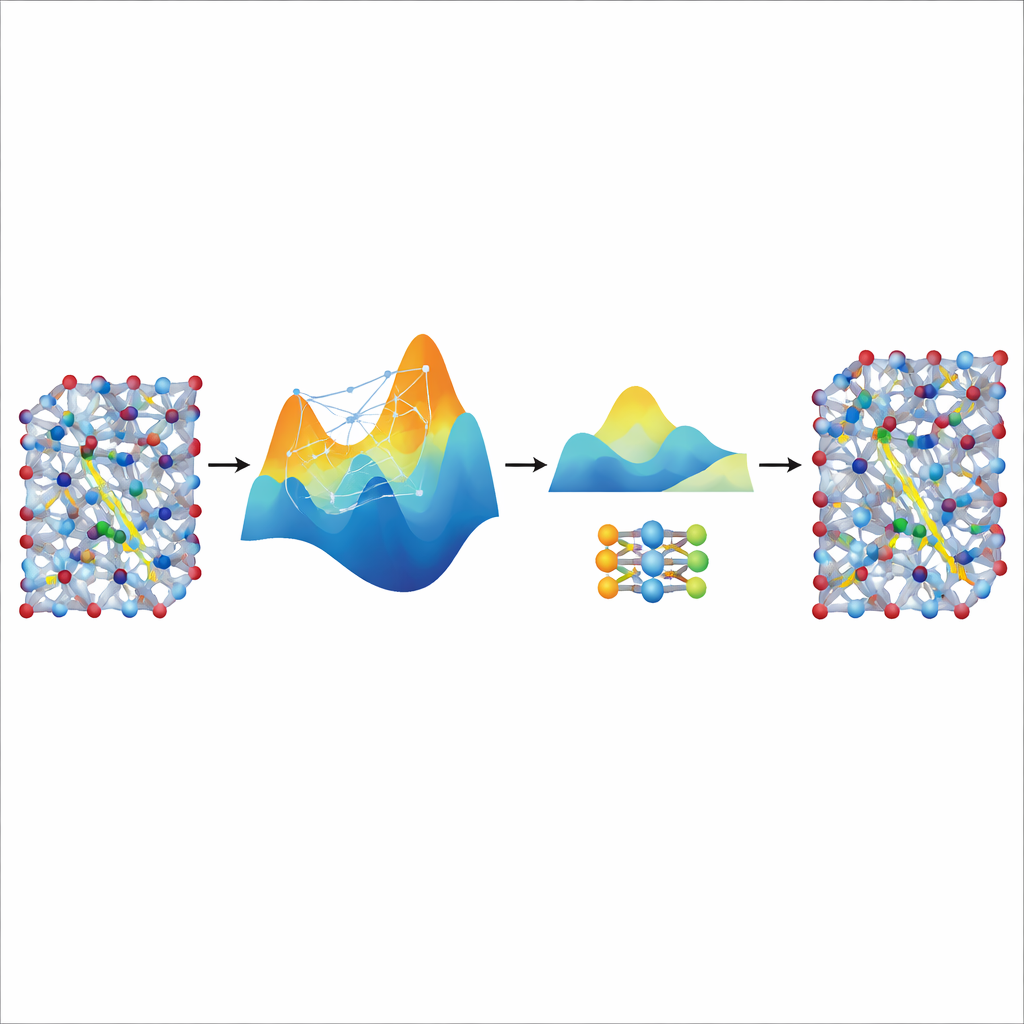
Snelheid destilleren uit een grote leraar
Hoewel het fijn-afgestelde MACE-model nauwkeurig en stabiel is, blijft het relatief zwaar en traag voor de echt lange en grote simulaties die nodig zijn om ionentransport in realistische batterijmaterialen te bestuderen. Om dit op te lossen gebruiken de auteurs het als een “leraar” voor een veel kleiner, lichtgewicht model bekend als NEP. Ze laten het afgestemde MACE-model extra synthetische trainingsdata genereren—duizenden atomaire configuraties gelabeld met de door het model voorspelde energieën en krachten—zonder extra kwantumberekeningen. Het trainen van NEP op deze gedistilleerde dataset produceert een compact model dat ongeveer twintig keer sneller draait en toch de voorspellingen van de leraar nauwkeurig nabootst. In grote supercel-simulaties reproduceert het gedistilleerde NEP-model sleutelkenmerken zoals superionische overgangen en geleidbaarheden bij kamertemperatuur die goed overeenkomen met experimenten.
Wat dit betekent voor toekomstige materialen
De studie demonstreert een praktisch recept voor het bouwen van betrouwbare, snelle machine-learning krachtvelden met slechts enkele honderden dure kwantumberekeningen: breed sampelen met een universeel model, het zorgvuldig finetunen en vervolgens het overdragen van die kennis naar een slankere student. Voor vaste-stof elektrolyten maakt deze benadering lange, grootschalige simulaties mogelijk die rechtstreeks vastleggen hoe lithiumionen zich een weg banen door complexe kristalstructuren, en realistische geleidbaarheden leveren in plaats van ruwe schattingen. Meer algemeen kan dezelfde workflow het ontwerp van veel functionele materialen versnellen en de droom van routine, hoog-fideliteits atomistische simulatie dichter bij alledaags onderzoek brengen.
Bronvermelding: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Trefwoorden: vaste-stof elektrolyten, machine learning potentialen, moleculaire dynamica, batterijmaterialen, materials simulatie