Clear Sky Science · de
Konstruktion von machine-learning-Interaktionspotenzialen mit minimaler Menge an ab initio-Daten
Klügere Simulationen für bessere Batterien
Festkörperbatterien versprechen sicherere Telefone, Autos und Netzspeicher, indem sie entzündliche flüssige Elektrolyte durch feste Materialien ersetzen, die Lithiumionen leiten. Die Suche und Prüfung neuer Festleiter ist jedoch langsam und teuer, insbesondere wenn Forschende auf aufwändige Supercomputer-Berechnungen angewiesen sind, die jedes Elektron nachverfolgen. Diese Arbeit zeigt, wie moderne Maschinenlernverfahren die Kosten drastisch senken können: Die Autorinnen und Autoren bauen genaue, schnelle „digitale Zwillinge“ der atomaren Kräfte mithilfe nur weniger hundert teurer Rechnungen statt Zehntausender und ebnen so den Weg für eine schnelle Durchmusterung von Batteriematerialien der nächsten Generation.
Warum es so schwierig ist, Atome zu simulieren
Um zu beurteilen, ob ein Feststoff Lithiumionen schnell transportiert, greifen Wissenschaftlerinnen und Wissenschaftler häufig auf ab initio-Molekulardynamik zurück, eine Goldstandardtechnik, die die atomare Bewegung aus der Quantenmechanik berechnet. Das Problem ist, dass sie so rechenintensiv ist, dass sie nicht routinemäßig für große Systeme oder lange Zeiten verwendet werden kann. Machine-Learning-Interaktionspotenziale bieten eine Abkürzung: Einmal trainiert, ahmen sie die zugrunde liegenden Quantenkräfte zu einem Bruchteil der Kosten nach. Der Aufbau solcher Modelle für ein spezifisches Material erforderte traditionell jedoch aufwändige „Active-Learning“-Schleifen und Tausende bis Zehntausende von Quantenrechnungen, was ihre breite Anwendung stark einschränkt.
Ein großes allgemeines Modell als Wegweiser nutzen
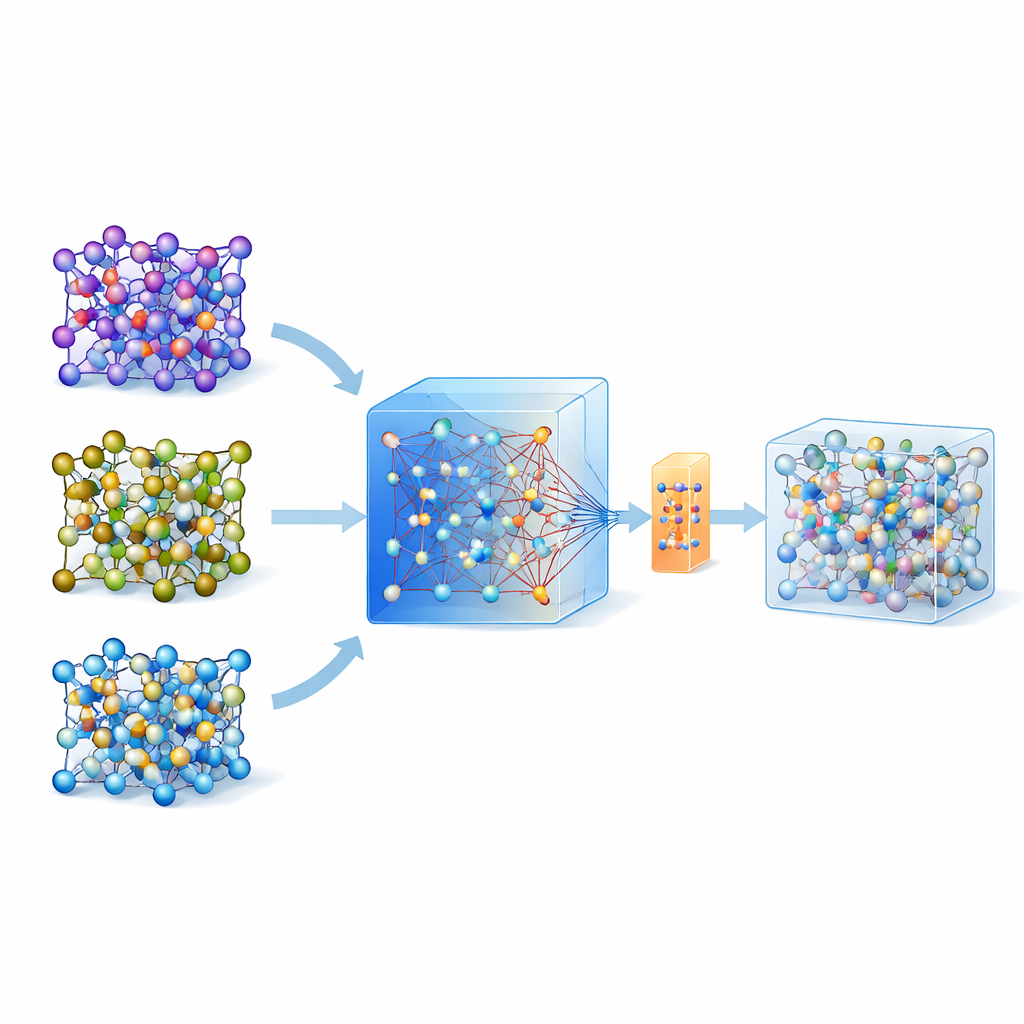
In den letzten Jahren sind universelle, große ML-Modelle entstanden, die auf umfangreichen Datenbanken von Quantenrechnungen über viele Materialien hinweg trainiert wurden. Eines dieser Modelle, genannt MACE-MP-0, dient hier als Ausgangspunkt. Die Autorinnen und Autoren testeten dieses universelle Modell zunächst an drei technologisch wichtigen Festkörper-Elektrolyten mit unterschiedlichen Chemien: einem Sulfid (LGPS), einem Oxid (LATP) und einem Halogenid (Li3YCl6). Sie stellten fest, dass MACE-MP-0 zwar die atomaren Trajektorien teils grob reproduzieren konnte, aber empfindliche Eigenschaften wie Lithium-Migrationsbarrieren und Diffusionsraten nicht genau genug vorhersagte. Dennoch stimmten seine Bewegungen durch den Konfigurationsraum eng mit den hochrangigen Rechnungen überein, wodurch es sich als exzellenter, preiswerter „Sampler“ relevanter atomarer Strukturen erwies.
Accurate Modelle aus winzigen Datensätzen bauen
Anstatt ein Modell in vielen Active-Learning-Runden wiederholt zu aktualisieren, schlagen die Autorinnen und Autoren eine Ein-Schuss-Strategie vor. Zuerst führen sie Hochtemperatur-Molekulardynamik mit dem universellen MACE-Modell durch, um viele atomare Schnappschüsse zu erzeugen. Danach wenden sie eine intelligente Resampling-Methode an, um nur etwa 200 besonders informative Konfigurationen auszuwählen und deren Energien und Kräfte mit vollständigen Quantenmethoden zu berechnen. Anstatt ein neues Modell von Grund auf zu trainieren, feinjustieren sie das vorhandene MACE-Modell an diesem kleinen, aber sorgfältig ausgewählten Datensatz, wobei sie sowohl konventionelle Updates als auch eine parameter-effiziente Variante namens ELoRA verwenden. Dieses getunte Modell wird nicht nur deutlich genauer bei Energiebarrieren und Diffusion, es übernimmt auch die dynamische Stabilität des ursprünglichen großen Modells und vermeidet unphysikalische atomare Zusammenbrüche, die bei von sehr begrenzten Daten trainierten Modellen oft auftreten.
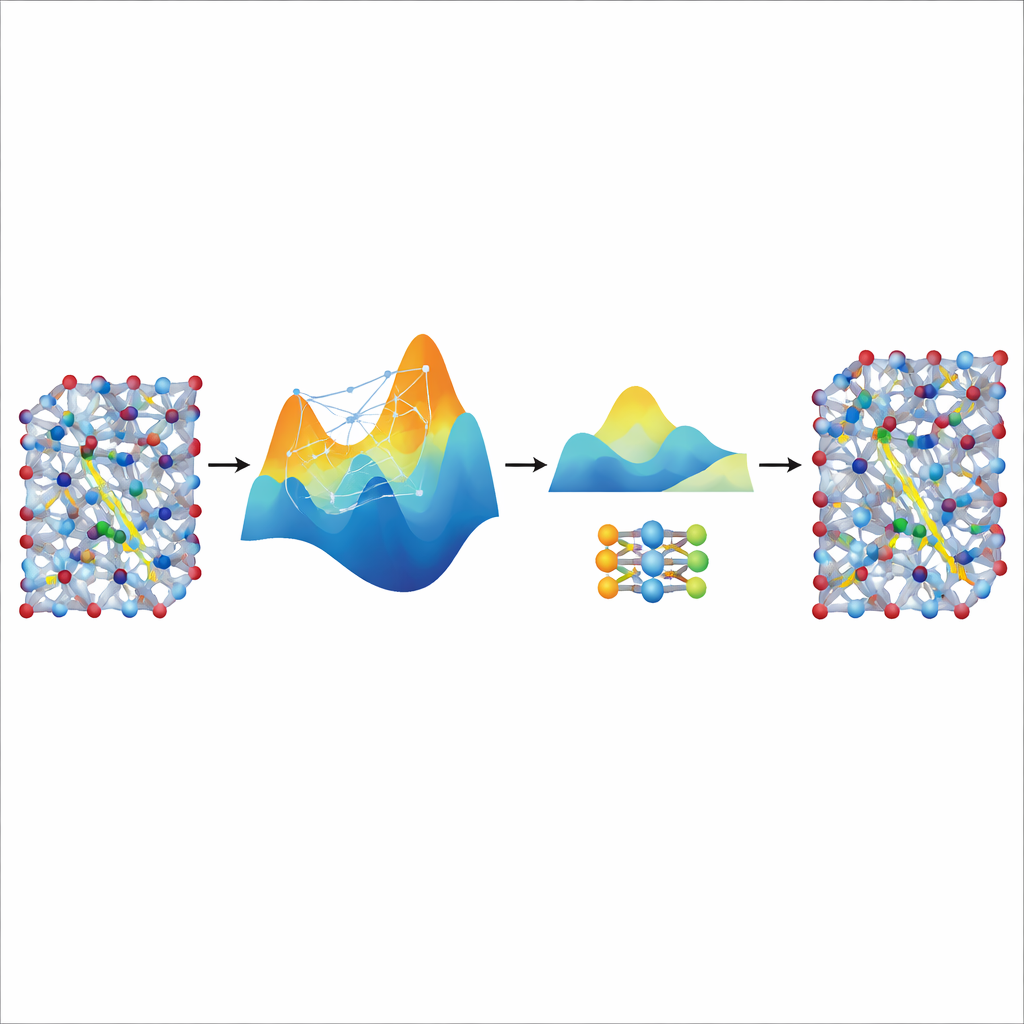
Geschwindigkeit aus einem großen Lehrer destillieren
Obwohl das feinabgestimmte MACE-Modell genau und stabil ist, bleibt es relativ schwergewichtig und langsam für die wirklich langen und großen Simulationen, die nötig sind, um Ionentransport in realistischen Batteriematerialien zu untersuchen. Um das zu lösen, nutzen die Autorinnen und Autoren es als „Lehrer“ für ein viel kleineres, leichtgewichtiges Modell namens NEP. Sie lassen das getunte MACE-Modell zusätzliche synthetische Trainingsdaten erzeugen—Tausende atomarer Konfigurationen, beschriftet mit seinen vorhergesagten Energien und Kräften—ohne weitere Quantenberechnungen. Das Training von NEP an diesem destillierten Datensatz ergibt ein kompaktes Modell, das etwa zwanzigmal schneller läuft und dennoch die Vorhersagen des Lehrers eng nachbildet. In großen Superzellen-Simulationen reproduziert das destillierte NEP-Modell Schlüsselfunktionen wie superionische Übergänge und Leitfähigkeiten bei Raumtemperatur, die gut mit experimentellen Befunden übereinstimmen.
Was das für zukünftige Materialien bedeutet
Die Studie demonstriert ein praxisnahes Rezept zum Aufbau zuverlässiger, schneller machine-learning-Kraftfelder mit nur wenigen hundert teuren Quantenberechnungen: weitreichend mit einem universellen Modell sampeln, es sorgfältig feinjustieren und seine Kenntnisse in einen schlankeren Schüler destillieren. Für Festkörper-Elektrolyte ermöglicht dieser Ansatz lange, großskalige Simulationen, die direkt erfassen, wie Lithiumionen durch komplexe Kristallstrukturen wandern, und realistische Leitfähigkeiten liefern statt grober Schätzungen. Allgemeiner könnte derselbe Workflow das Design vieler funktionaler Materialien beschleunigen und den Traum von routinemäßigen, hochgenauen atomistischen Simulationen deutlich näher an die tägliche Forschungspraxis bringen.
Zitation: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Schlüsselwörter: Festkörper-Elektrolyte, Machine-Learning-Potenziale, Molekulardynamik, Batteriematerialien, Materialsimulation