Clear Sky Science · pt
Construindo potenciais interatômicos por aprendizado de máquina com a quantidade mínima de dados ab initio
Simulações mais inteligentes para baterias melhores
Baterias de estado sólido prometem telefones, carros e armazenamento de rede mais seguros ao substituir eletrólitos líquidos inflamáveis por materiais sólidos que conduzem íons de lítio. Mas encontrar e testar novos condutores sólidos é lento e caro, especialmente quando os pesquisadores dependem de cálculos pesados em supercomputadores que acompanham cada elétron. Este artigo mostra como usar aprendizado de máquina moderno para reduzir dramaticamente esse custo: os autores constroem “gêmeos digitais” rápidos e precisos das forças atômicas usando apenas algumas centenas de cálculos caros em vez de dezenas de milhares, abrindo caminho para triagens rápidas de materiais para a próxima geração de baterias.
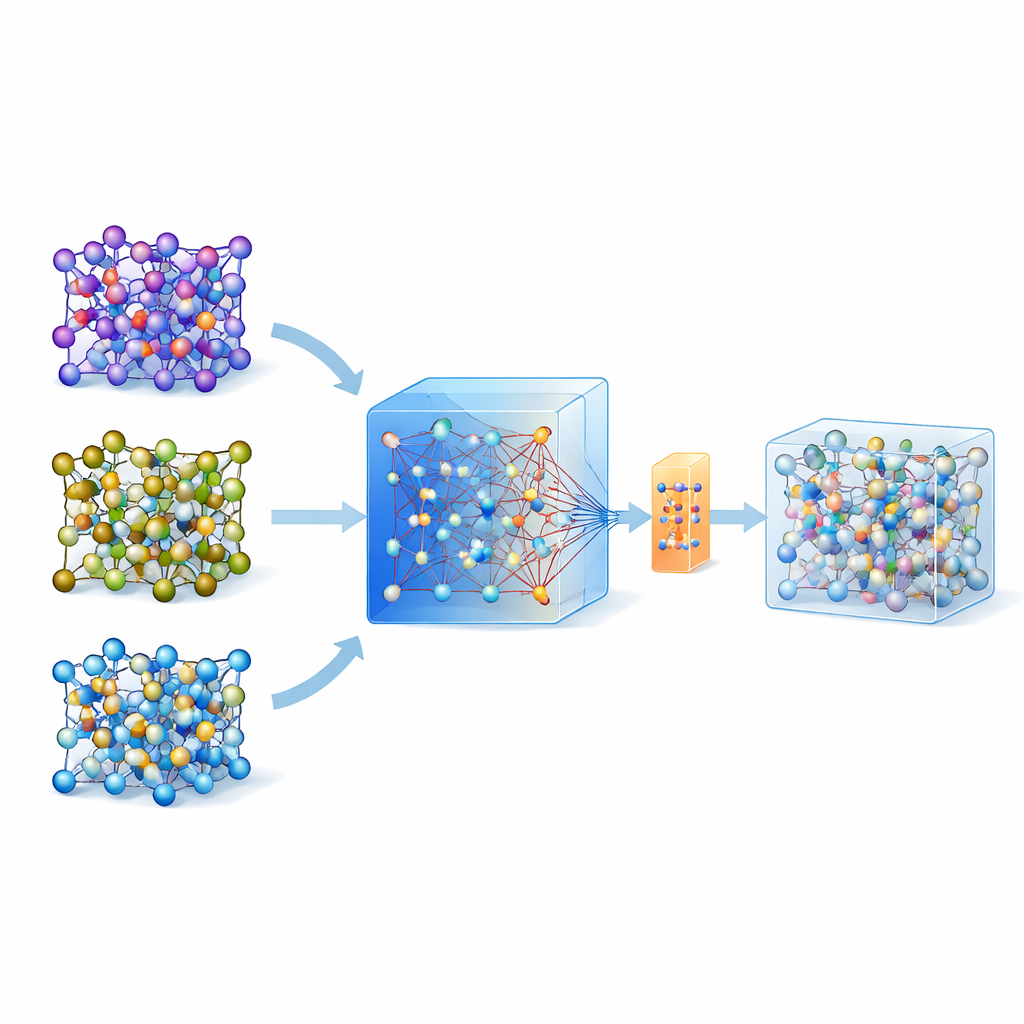
Por que simular átomos é tão difícil
Para avaliar se um material sólido transportará íons de lítio rapidamente, os cientistas frequentemente recorrem à dinâmica molecular ab initio, uma técnica padrão-ouro que computa o movimento atômico a partir da mecânica quântica. O problema é que ela é tão exigente computacionalmente que não pode ser usada rotineiramente para sistemas grandes ou tempos longos. Potenciais interatômicos por aprendizado de máquina oferecem um atalho: uma vez treinados, eles imitam as forças quânticas subjacentes a uma fração do custo. No entanto, construir tais modelos para um material específico tradicionalmente exigia laços intrincados de “aprendizado ativo” e milhares a dezenas de milhares de cálculos quânticos, o que limita muito sua aplicação.
Usando um grande modelo geral como guia
Nos últimos anos surgiram modelos universais e grandes treinados em enormes bancos de dados de cálculos quânticos cobrindo muitos materiais. Um desses modelos, chamado MACE-MP-0, serve como ponto de partida aqui. Os autores primeiro testaram esse modelo universal em três eletrólitos de estado sólido tecnologicamente importantes que abrangem químicas diferentes: um sulfeto (LGPS), um óxido (LATP) e um haleto (Li3YCl6). Eles constataram que, embora o MACE-MP-0 pudesse reproduzir grosseiramente as trajetórias atômicas das simulações de referência caras, ele não previa propriedades delicadas como barreiras de migração do lítio e taxas de difusão com precisão suficiente. Ainda assim, sua exploração do espaço de configurações atômicas correspondia de perto aos cálculos de alto nível, tornando-o um excelente e barato “amostrador” de estruturas atômicas relevantes.
Construindo modelos precisos a partir de conjuntos de dados mínimos
Em vez de atualizar repetidamente um modelo com muitas rodadas de aprendizado ativo, os autores propõem uma estratégia de tiro único. Primeiro, eles executam dinâmica molecular em alta temperatura usando o modelo universal MACE para gerar muitas capturas atômicas. Em seguida, aplicam um método inteligente de reamostragem para escolher apenas cerca de 200 configurações especialmente informativas e calculam suas energias e forças usando métodos quânticos completos. Em vez de treinar um modelo novo a partir do zero, eles afinam o modelo MACE existente nesse conjunto de dados pequeno, porém cuidadosamente selecionado, usando tanto atualização convencional quanto uma variante eficiente em parâmetros chamada ELoRA. Esse modelo ajustado não só se torna significativamente mais preciso para barreiras de energia e difusão, como também herda a estabilidade dinâmica do grande modelo original, evitando colapsos atômicos não físicos que costumam afetar modelos treinados com dados muito limitados.
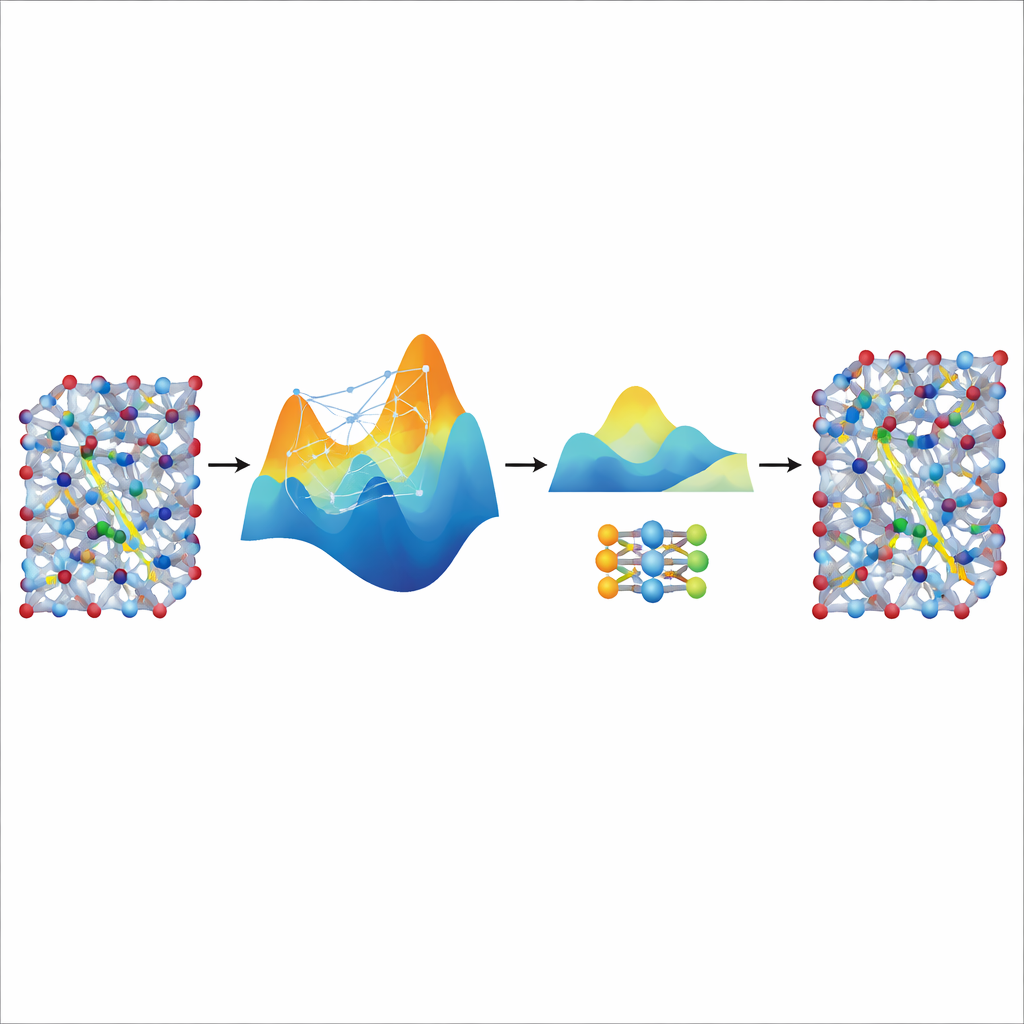
Destilando velocidade de um grande professor
Embora o modelo MACE afinado seja preciso e estável, ele continua relativamente pesado e lento para as simulações realmente longas e grandes necessárias para estudar o transporte iônico em materiais de bateria realistas. Para resolver isso, os autores o usam como “professor” para um modelo muito menor e leve conhecido como NEP. Eles deixam o modelo MACE ajustado gerar dados sintéticos adicionais de treinamento—milhares de configurações atômicas rotuladas com suas energias e forças previstas—sem cálculos quânticos extras. Treinar o NEP nesse conjunto destilado produz um modelo compacto que roda cerca de vinte vezes mais rápido ao reproduzir de perto as previsões do professor. Em grandes simulações com supercélulas, o modelo NEP destilado reproduce características-chave como transições superiônicas e condutividades à temperatura ambiente que se alinham bem com experimentos.
O que isso significa para materiais futuros
O estudo demonstra uma receita prática para construir campos de forças por aprendizado de máquina confiáveis e rápidos usando apenas algumas centenas de cálculos quânticos caros: amostrar amplamente com um modelo universal, afiná‑lo com cuidado e então destilar seu conhecimento em um aluno mais enxuto. Para eletrólitos de estado sólido, essa abordagem permite simulações longas e em larga escala que capturam diretamente como íons de lítio se deslocam por estruturas cristalinas complexas, fornecendo condutividades realistas em vez de estimativas grosseiras. Mais amplamente, o mesmo fluxo de trabalho pode acelerar o projeto de muitos materiais funcionais, aproximando o sonho de simulação atomística rotineira e de alta fidelidade da prática diária de pesquisa.
Citação: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Palavras-chave: eletrólitos de estado sólido, potenciais por aprendizado de máquina, dynamics molecular, materiais para baterias, simulação de materiais