Clear Sky Science · fr
Construction de potentiels interatomiques par apprentissage automatique avec un nombre minimal de données ab initio
Des simulations plus intelligentes pour de meilleures batteries
Les batteries à l'état solide promettent des téléphones, des voitures et du stockage réseau plus sûrs en remplaçant les électrolytes liquides inflammables par des matériaux solides conducteurs d'ions lithium. Mais découvrir et tester de nouveaux conducteurs solides reste long et coûteux, surtout quand les chercheurs s'appuient sur des calculs intensifs sur supercalculateurs qui suivent chaque électron. Cet article montre comment utiliser l'apprentissage automatique moderne pour réduire drastiquement ce coût : les auteurs construisent des « jumeaux numériques » rapides et précis des forces atomiques en n'utilisant que quelques centaines de calculs coûteux au lieu de dizaines de milliers, ouvrant la voie au criblage rapide des matériaux de batterie de nouvelle génération.
Pourquoi simuler des atomes est si difficile
Pour juger si un matériau solide transporte rapidement des ions lithium, les scientifiques recourent souvent à la dynamique moléculaire ab initio, une technique de référence qui calcule le mouvement atomique à partir de la mécanique quantique. Le problème est qu'elle est si exigeante en calcul qu'on ne peut pas l'utiliser systématiquement pour de grands systèmes ou de longues durées. Les potentiels interatomiques par apprentissage automatique offrent une raccourci : une fois entraînés, ils imitent les forces quantiques sous-jacentes à une fraction du coût. Toutefois, construire de tels modèles pour un matériau spécifique a traditionnellement nécessité des boucles d'« apprentissage actif » complexes et des milliers à des dizaines de milliers de calculs quantiques, ce qui limite fortement leur déploiement.
Utiliser un grand modèle général comme guide
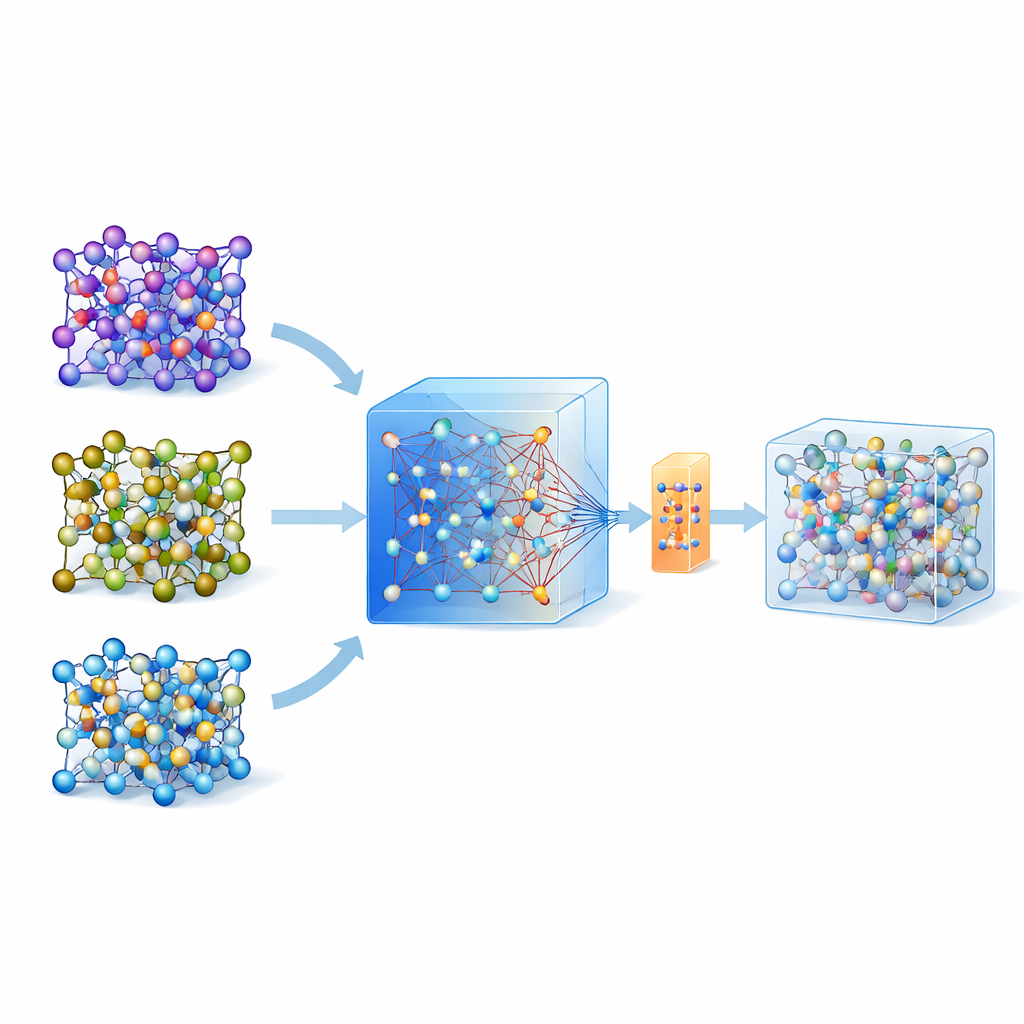
Ces dernières années ont vu l'émergence de modèles universels et volumineux entraînés sur d'énormes bases de données de calculs quantiques couvrant de nombreux matériaux. L'un de ces modèles, appelé MACE-MP-0, sert ici de point de départ. Les auteurs ont d'abord testé ce modèle universel sur trois électrolytes solides technologiquement importants couvrant différentes chimies : un sulfure (LGPS), un oxyde (LATP) et un halogénure (Li3YCl6). Ils ont constaté que, bien que MACE-MP-0 puisse reproduire grossièrement les trajectoires atomiques issues des simulations de référence coûteuses, il ne prédisait pas avec suffisamment de précision des propriétés délicates comme les barrières de migration du lithium et les taux de diffusion. Néanmoins, son exploration de l'espace des configurations atomiques correspondait étroitement aux calculs de haut niveau, ce qui en fait un excellent « échantillonneur » peu coûteux des structures atomiques pertinentes.
Construire des modèles précis à partir de jeux de données minuscules
Plutôt que de mettre à jour un modèle à plusieurs reprises avec de nombreuses itérations d'apprentissage actif, les auteurs proposent une stratégie en une seule étape. Ils exécutent d'abord des dynamiques moléculaires à haute température en utilisant le modèle universel MACE pour générer de nombreuses instantanés atomiques. Ils appliquent ensuite une méthode de rééchantillonnage intelligente pour ne sélectionner qu'environ 200 configurations particulièrement informatives et calculer leurs énergies et forces avec des méthodes quantiques complètes. Plutôt que d'entraîner un modèle neuf depuis zéro, ils adaptent (fine-tune) le modèle MACE existant sur cet ensemble de données petit mais soigneusement choisi, en utilisant à la fois une mise à jour conventionnelle et une variante économe en paramètres appelée ELoRA. Ce modèle ajusté devient non seulement beaucoup plus précis pour les barrières énergétiques et la diffusion, mais il hérite aussi de la stabilité dynamique du grand modèle initial, évitant les collapsus atomiques non physiques qui affectent souvent les modèles entraînés sur des données très limitées.
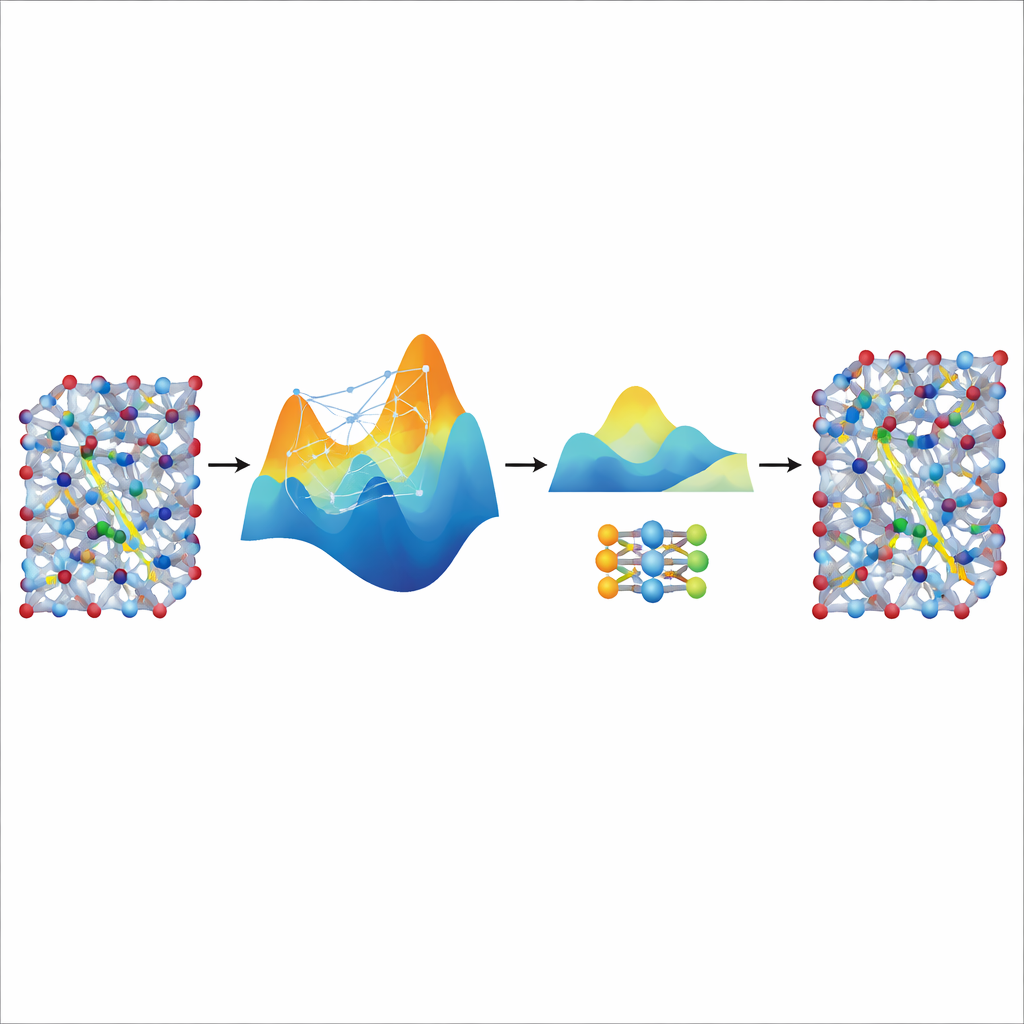
Extraire la rapidité d'un grand enseignant
Bien que le modèle MACE ajusté soit précis et stable, il reste relativement lourd et lent pour les simulations très longues et volumineuses nécessaires à l'étude du transport d'ions dans des matériaux de batterie réalistes. Pour y remédier, les auteurs l'utilisent comme « enseignant » pour un modèle beaucoup plus petit et léger connu sous le nom de NEP. Ils laissent le modèle MACE ajusté générer des données d'entraînement synthétiques supplémentaires — des milliers de configurations atomiques étiquetées par ses prédictions d'énergies et de forces — sans calculs quantiques supplémentaires. En entraînant NEP sur ce jeu de données distillé, on obtient un modèle compact qui fonctionne environ vingt fois plus vite tout en reproduisant fidèlement les prédictions de l'enseignant. Dans de grandes simulations en supercellules, le modèle NEP distillé reproduit des caractéristiques clés telles que les transitions superioniques et les conductivités à température ambiante qui concordent bien avec les expériences.
Ce que cela signifie pour les matériaux futurs
L'étude démontre une recette pratique pour construire des champs de forces d'apprentissage automatique fiables et rapides en n'utilisant que quelques centaines de calculs quantiques coûteux : échantillonner largement avec un modèle universel, l'affiner soigneusement, puis distiller son savoir dans un étudiant plus léger. Pour les électrolytes à l'état solide, cette approche permet des simulations longues et à grande échelle qui capturent directement la manière dont les ions lithium traversent des structures cristallines complexes, fournissant des conductivités réalistes plutôt que des estimations grossières. Plus généralement, le même flux de travail pourrait accélérer la conception de nombreux matériaux fonctionnels, rapprochant le rêve de simulations atomistiques de haute fidélité et d'utilisation courante dans la recherche quotidienne.
Citation: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Mots-clés: électrolytes à l'état solide, potentiels d'apprentissage automatique, dynamique moléculaire, matériaux pour batteries, simulation de matériaux