Clear Sky Science · en
Constructing machine learning interatomic potentials with minimum amount of ab initio data
Smarter Simulations for Better Batteries
Solid-state batteries promise safer phones, cars, and grid storage by replacing flammable liquid electrolytes with solid materials that conduct lithium ions. But finding and testing new solid conductors is slow and expensive, especially when researchers rely on heavy supercomputer calculations that track every electron. This paper shows how to use modern machine learning to dramatically cut that cost: the authors build accurate, fast “digital twins” of atomic forces using only a few hundred expensive calculations instead of tens of thousands, opening the door to rapid screening of next-generation battery materials.
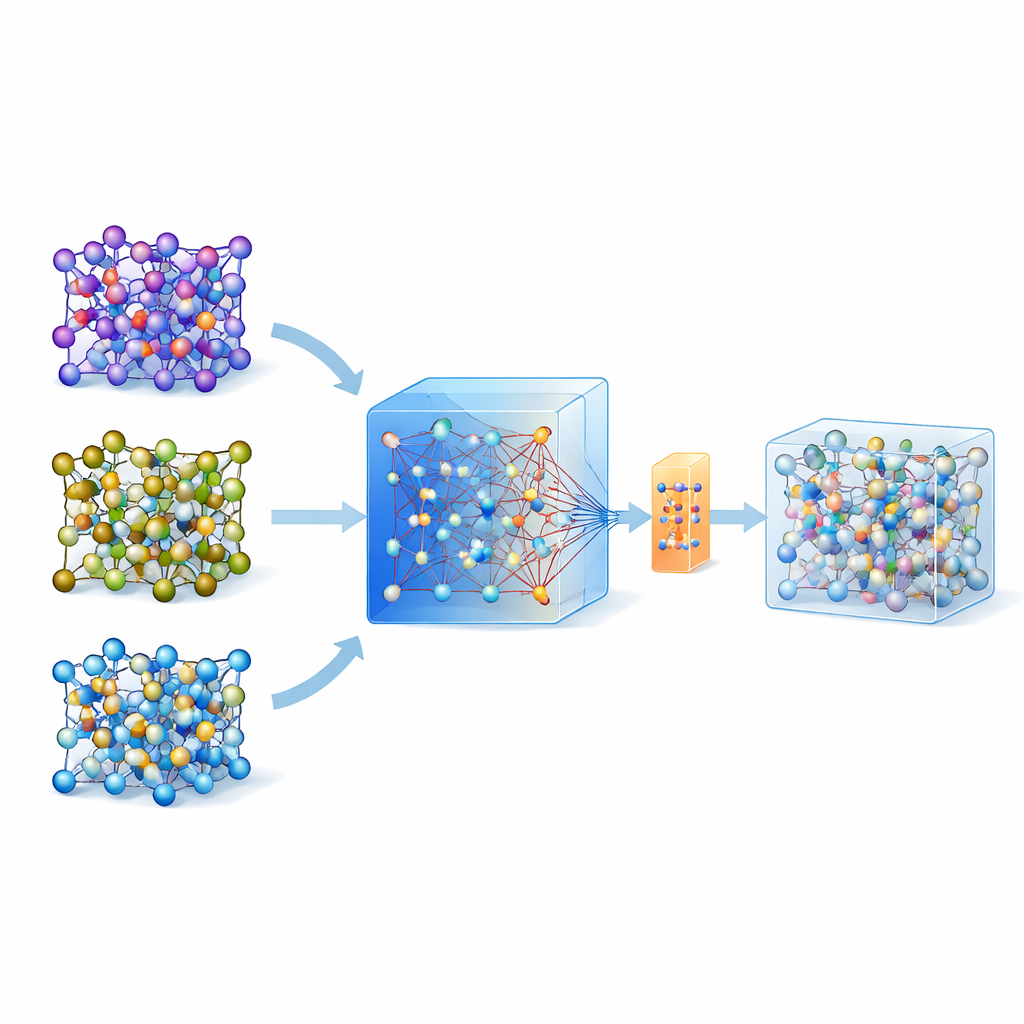
Why Simulating Atoms Is So Hard
To judge whether a solid material will carry lithium ions quickly, scientists often turn to ab initio molecular dynamics, a gold-standard technique that computes atomic motion from quantum mechanics. The catch is that it is so computationally demanding that it cannot be used routinely for large systems or long times. Machine learning interatomic potentials offer a shortcut: once trained, they imitate the underlying quantum forces at a fraction of the cost. However, building such models for a specific material has traditionally required intricate “active learning” loops and thousands to tens of thousands of quantum calculations, which greatly limits how widely they can be deployed.
Using a Big General Model as a Guide
Recent years have seen the rise of universal, large machine-learning models trained on huge databases of quantum calculations across many materials. One such model, called MACE-MP-0, serves as the starting point here. The authors first tested this universal model on three technologically important solid-state electrolytes that span different chemistries: a sulfide (LGPS), an oxide (LATP), and a halide (Li3YCl6). They found that, while MACE-MP-0 could roughly reproduce the atomic trajectories from expensive reference simulations, it did not predict delicate properties such as lithium migration barriers and diffusion rates accurately enough. Still, its motion through atomic configuration space closely matched the high-level calculations, making it an excellent, cheap “sampler” of relevant atomic structures.
Building Accurate Models from Tiny Datasets
Instead of repeatedly updating a model with many rounds of active learning, the authors propose a single-shot strategy. First, they run high-temperature molecular dynamics using the universal MACE model to generate many atomic snapshots. Then they apply a smart resampling method to pick only about 200 especially informative configurations and calculate their energies and forces using full quantum methods. Rather than training a fresh model from scratch, they fine-tune the existing MACE model on this small but carefully chosen dataset, using both conventional updating and a parameter-efficient variant called ELoRA. This tuned model not only becomes significantly more accurate for energy barriers and diffusion, it also inherits the dynamical stability of the original large model, avoiding unphysical atomic collapses that often plague models trained from very limited data.
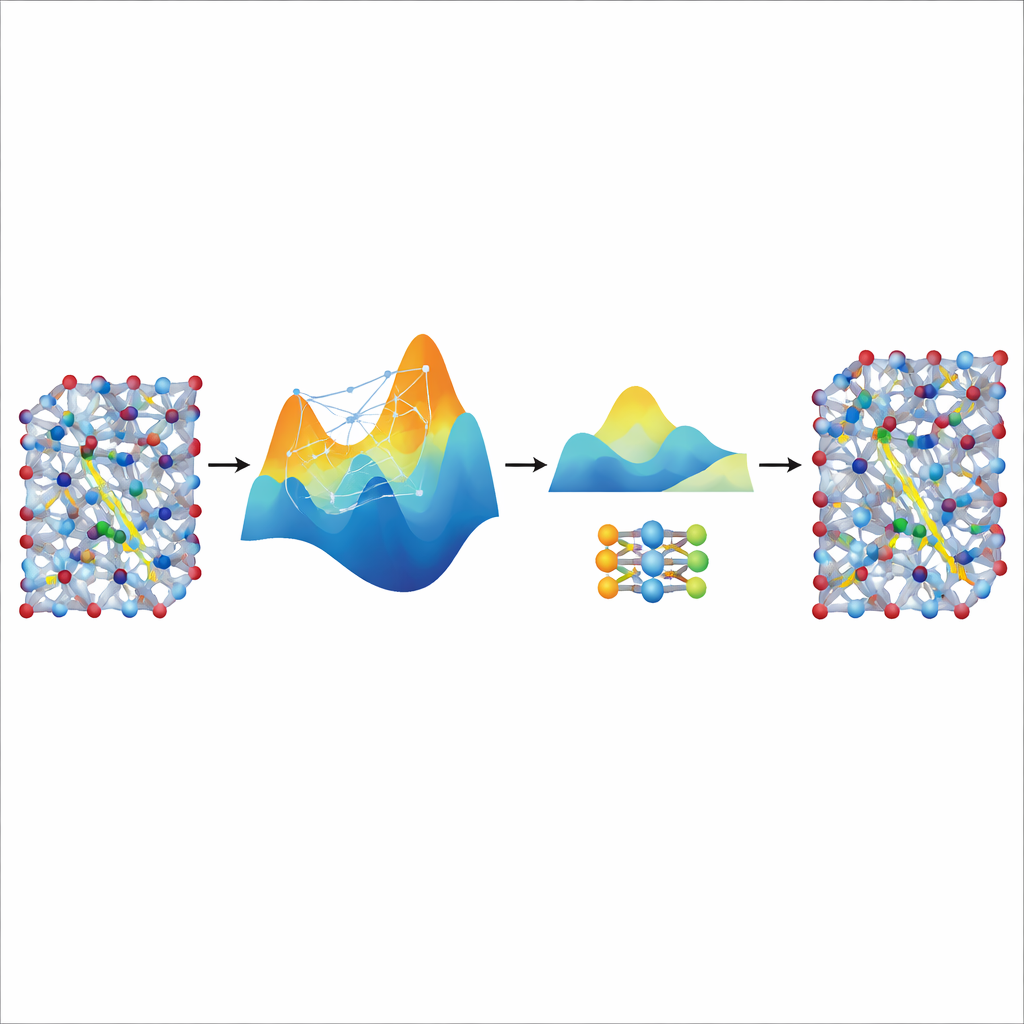
Distilling Speed from a Large Teacher
Although the fine-tuned MACE model is accurate and stable, it remains relatively heavy and slow for the truly long and large simulations needed to study ion transport in realistic battery materials. To solve this, the authors use it as a “teacher” for a much smaller, lightweight model known as NEP. They let the tuned MACE model generate additional synthetic training data—thousands of atomic configurations labeled with its predicted energies and forces—without any extra quantum calculations. Training NEP on this distilled dataset produces a compact model that runs about twenty times faster while closely matching the teacher’s predictions. In large supercell simulations, the distilled NEP model reproduces key features such as superionic transitions and room-temperature conductivities that align well with experiments.
What This Means for Future Materials
The study demonstrates a practical recipe for building reliable, fast machine-learning force fields using only a few hundred expensive quantum calculations: sample widely with a universal model, fine-tune it carefully, and then distill its knowledge into a leaner student. For solid-state electrolytes, this approach enables long, large-scale simulations that directly capture how lithium ions weave through complex crystal structures, providing realistic conductivities instead of crude estimates. More broadly, the same workflow could accelerate the design of many functional materials, bringing the dream of routine, high-fidelity atomistic simulation much closer to everyday research practice.
Citation: Zhang, W., Wu, X., Wang, C. et al. Constructing machine learning interatomic potentials with minimum amount of ab initio data. npj Comput Mater 12, 174 (2026). https://doi.org/10.1038/s41524-026-02023-y
Keywords: solid-state electrolytes, machine learning potentials, molecular dynamics, battery materials, materials simulation