Clear Sky Science · zh
序列性转录波与NF-κB驱动的染色质重塑指导药物诱导的癌细胞去分化
为什么癌细胞在治疗后会反弹
现代抗癌药物可以显著缩小肿瘤,但疾病常常会复发。该研究探索了癌细胞利用的一条隐藏逃逸路线:部分细胞并非被永久杀死或通过突变获益,而是暂时“回退”到更原始、类干细胞的状态,从而帮助其在治疗中存活。通过在基因和染色质(DNA的包装方式)水平上随时间追踪这些变化,作者揭示了一个由感受压力的信号通路NF-κB/RelA控制的分步程序,使癌细胞进入并随后离开耐药状态。理解这一可逆过程为延长治疗有效期提供了新的思路。
通过变得不那么专化而存活的细胞
研究者将焦点放在由突变BRAF驱动的黑色素瘤上,这类肿瘤是常见的精准药物靶点。当他们用BRAF抑制剂处理高度可塑的黑色素瘤细胞时,细胞并非简单地死亡或保持不变。相反,它们经历了一系列身份变化:从产色素的黑色素瘤细胞,转为分裂缓慢且耐药的神经嵴样状态,最终进入更原始的类间充质状态,对多种治疗高度耐受。值得注意的是,撤药后这些细胞逐渐恢复到原来的、对药物敏感的身份,表明耐药并非由永久DNA突变引起,而是一种灵活的状态变化。
不是直线而是循环路径
为了解成千上万变化基因的含义,团队使用基于信息论的方法将数据压缩为仅两个主要的协调基因活动“波”。第一波为早期波,在治疗数日内启动,与染色质的快速变化和细胞分裂停滞相关;第二波为后期波,控制定义细胞身份和侵袭行为的基因。将这两波绘于坐标系上显示,进入耐药与退出耐药的路径并不重合:细胞在染色质构型中记住了其治疗历史,这种行为称为滞后性(hysteresis)。这一记忆意味着即便基因表达看似相似,底层的DNA包装也可能截然不同。
压力信号如何重接DNA包装
进一步挖掘发现,药物处理扰乱了细胞的抗氧化防御,导致活性氧(ROS)积累。这种氧化应激激活了NF-κB/RelA这一主控应激反应因子。RelA一旦被激活,进入细胞核并与两种染色质修饰酶KDM5B和HDAC1一同结合基因组的多个位点。在数百个靶基因处,包括关键的黑色素瘤身份基因如SOX10和MITF,这个三联体去除了激活性的组蛋白标记并使局部染色质可及性降低。基因表达下调并在药物存在期间保持低水平,将细胞锁定在慢周期、耐药的状态。当阻断RelA进入细胞核时,染色质标记得以恢复,SOX10回复,细胞再次对BRAF抑制剂敏感。
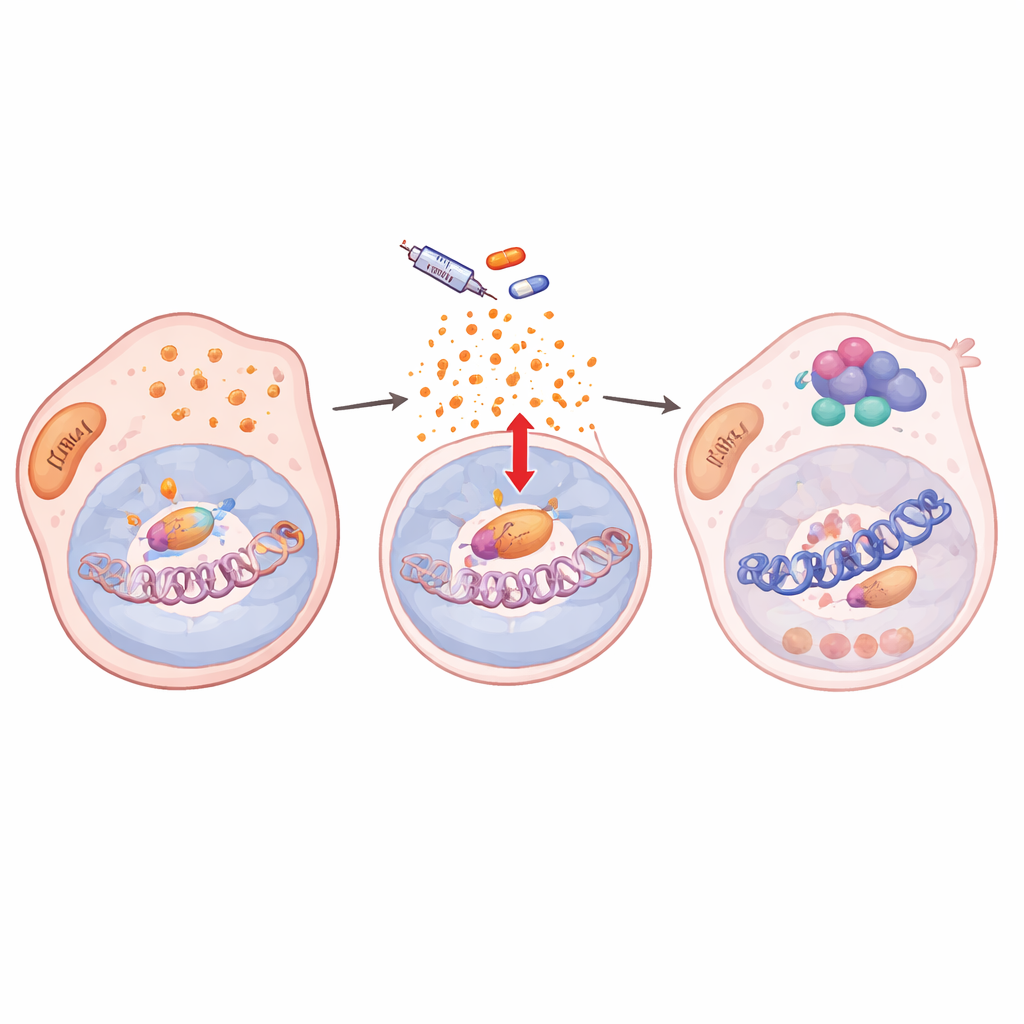
编码于染色质的可塑性,在多种癌症中共享
团队随后探讨了为何某些肿瘤比其他肿瘤更容易发生这种形态转变。在多条黑色素瘤细胞系中,他们发现药物诱导变化的程度与治疗前RelA靶基因处染色质的开放和活跃程度相关。具有更“宽松”染色质的细胞系表现出更大的转变,并且在将BRAF抑制剂与针对RelA或其染色质伙伴的药物联合使用时获益更明显。最后,他们证明在肺癌和结直肠癌中,针对各自驱动基因的治疗也通过类似的ROS–NF-κB–染色质通路驱动进入耐药持续细胞状态。在所有情形下,阻断NF-κB/RelA激活或降低ROS都能抑制持留细胞的出现与再生长。
这对未来癌症治疗意味着什么
这项工作将多种癌症中的药物耐受性重新阐释为一种可逆的应激反应程序,而非单纯的遗传问题。癌细胞感知治疗引发的氧化应激,激活NF-κB/RelA,并利用相关的染色质重塑酶暂时关闭维持其特化和药物敏感性的基因。由于该过程依赖于DNA的包装状态而非永久突变,因此有可能被干预。研究结果支持将靶向药物与阻断NF-κB/RelA信号或其染色质修饰伙伴的药物联合使用的治疗策略,尤其适用于那些染色质已被预设为易发生此类可塑性的肿瘤。通过阻止细胞首先进入持留状态,这类组合可能使缓解更深、更持久。
引用: Su, Y., Liu, C., Lu, X. et al. Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer. Nat Commun 17, 3228 (2026). https://doi.org/10.1038/s41467-026-71349-4
关键词: 耐药持续细胞, NF-kB RelA 信号通路, 染色质重塑, 癌细胞可塑性, 靶向治疗耐药