Clear Sky Science · fr
Vagues transcriptionnelles séquentielles et remodelage chromatinaire dirigé par NF-κB orientent la dédifférenciation induite par les médicaments dans le cancer
Pourquoi les cellules cancéreuses peuvent rebondir après un traitement
Les médicaments anticancéreux modernes peuvent réduire considérablement les tumeurs, mais la maladie revient trop souvent. Cette étude explore une voie d’évasion cachée utilisée par les cellules cancéreuses : au lieu d’être définitivement tuées ou de muter, certaines cellules « rembobinent » temporairement vers un état plus primitif, apparenté à des cellules souches, qui les aide à survivre au traitement. En suivant ces changements au fil du temps, tant au niveau des gènes que de la chromatine (la façon dont l’ADN est empaqueté), les auteurs mettent au jour un programme étape par étape, contrôlé par une voie de détection du stress appelée NF‑κB/RelA, qui permet aux cellules cancéreuses d’entrer puis de sortir d’un état tolérant aux médicaments. Comprendre ce processus réversible suggère de nouvelles manières de prolonger l’efficacité des traitements.
Des cellules qui survivent en devenant moins spécialisées
Les chercheurs se sont concentrés sur des mélanomes dirigés par BRAF mutant, une cible fréquente des médicaments de précision. Lorsqu’ils ont traité des cellules de mélanome très adaptables avec un inhibiteur de BRAF, les cellules n’ont pas simplement péri ni conservé leur état. Elles ont traversé une séquence d’identités : de cellules productrices de pigment, à un état similaire à la crête neurale qui se divise lentement et tolère les médicaments, puis à un état encore plus primitif, de type mésenchymateux, fortement résistant à de nombreuses thérapies. De façon remarquable, lorsque le médicament a été retiré, ces mêmes cellules sont progressivement revenues à leur identité d’origine, sensible au traitement, montrant que la résistance n’était pas due à des mutations d’ADN permanentes mais à un changement d’état réversible.
Un trajet en boucle plutôt qu’une ligne droite
Pour interpréter des milliers de gènes changeants, l’équipe a utilisé une méthode basée sur la théorie de l’information pour compresser les données en seulement deux grandes « vagues » d’activité génique coordonnée. La première, une vague précoce, s’est déclenchée en quelques jours de traitement et était liée à des changements rapides de la chromatine et à un arrêt de la division cellulaire. La seconde, plus tardive, contrôlait des gènes définissant l’identité cellulaire et le comportement invasif. En traçant ces deux vagues l’une par rapport à l’autre, ils ont révélé que le chemin entrant dans la tolérance aux médicaments et le chemin de sortie ne se chevauchaient pas : les cellules gardaient la mémoire de leur histoire de traitement dans la configuration de leur chromatine, un comportement connu sous le nom d’hystérésis. Cette mémoire signifie que, même lorsque l’activité génique semble similaire, l’empaquetage de l’ADN sous-jacent peut être très différent.
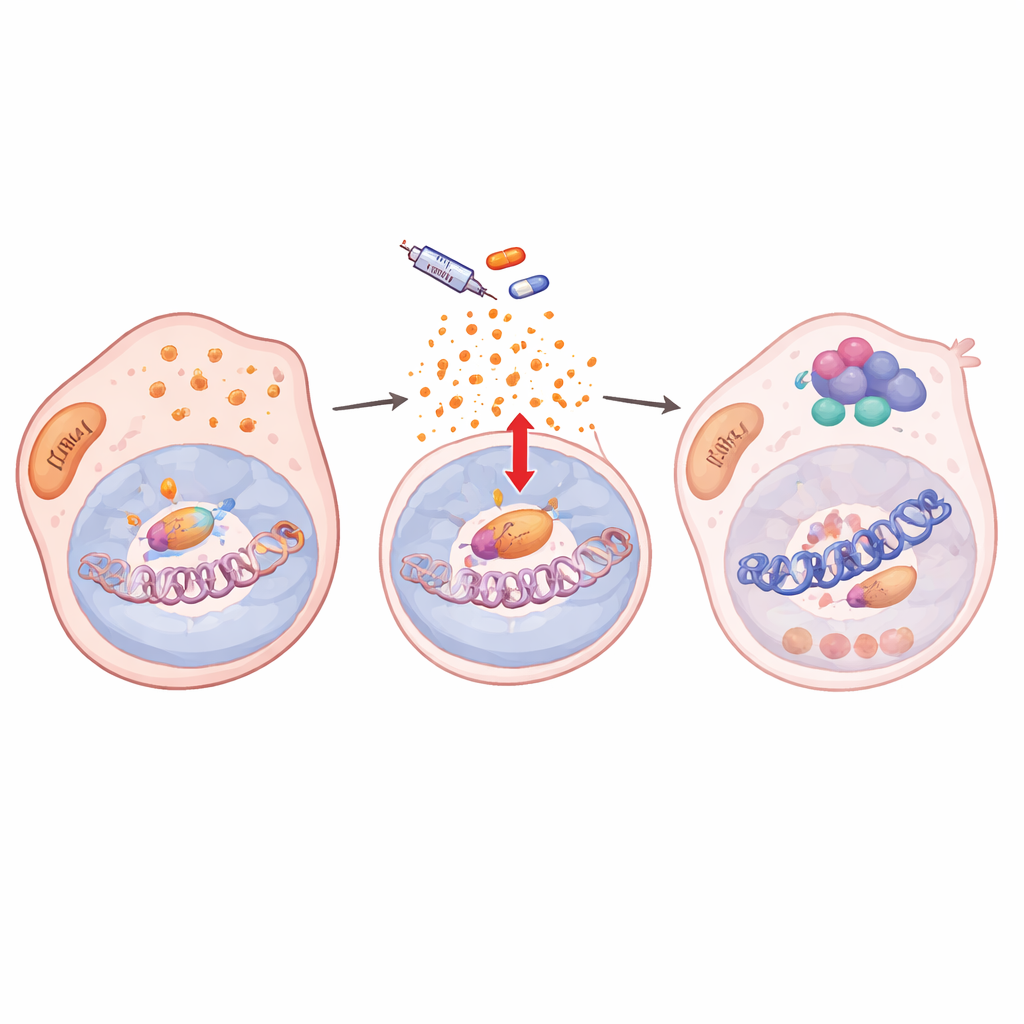
Comment les signaux de stress réorganisent l’empaquetage de l’ADN
Plus en profondeur, les auteurs ont constaté que le traitement perturbait les défenses antioxydantes des cellules, entraînant une accumulation d’espèces réactives de l’oxygène (ROS). Ce stress oxydatif activait NF‑κB/RelA, un facteur maître de la réponse au stress. Une fois activé, RelA migrait vers le noyau et se liait à de nombreux sites du génome en association avec deux enzymes modifiant la chromatine, KDM5B et HDAC1. Sur des centaines de gènes cibles, y compris des gènes clés de l’identité du mélanome tels que SOX10 et MITF, ce trio a retiré des marques d’histones activatrices et rendu la chromatine locale moins accessible. L’activité génique chutait et restait faible tant que le médicament était présent, verrouillant les cellules dans un état de cycle lent et tolérant au traitement. Lorsque l’entrée nucléaire de RelA était bloquée, les marques chromatiniques se rétablissaient, SOX10 rebondissait et les cellules redevenaient sensibles à l’inhibition de BRAF.
Une plasticité encodée dans la chromatine, partagée entre différents cancers
L’équipe s’est ensuite demandé pourquoi certaines tumeurs sont plus susceptibles que d’autres à ce comportement de métamorphose. À travers plusieurs lignées cellulaires de mélanome, ils ont trouvé que l’ampleur du changement induit par le médicament corrélait avec le degré d’ouverture et d’activité de la chromatine au niveau des gènes cibles de RelA avant le traitement. Les lignées présentant une chromatine plus « permissive » montraient des transitions plus larges et bénéficiaient davantage de la combinaison d’inhibiteurs de BRAF avec des médicaments ciblant RelA ou ses partenaires chromatiniques. Enfin, ils ont montré qu’une voie ROS–NF‑κB–chromatine similaire pilote l’entrée dans des états persistants tolérants aux médicaments dans les cancers du poumon et du côlon traités contre leurs oncogènes moteurs. Dans tous les cas, bloquer l’activation de NF‑κB/RelA ou atténuer les ROS réduisait l’émergence et la repousse des cellules persistantes.
Ce que cela signifie pour les traitements futurs du cancer
Ce travail requalifie la résistance aux médicaments dans de nombreux cancers en tant que programme de réponse au stress réversible plutôt que comme un problème purement génétique. Les cellules cancéreuses détectent le stress oxydatif induit par la thérapie, activent NF‑κB/RelA et utilisent des remodelleurs de chromatine associés pour éteindre temporairement les gènes qui les maintiennent spécialisées et sensibles aux médicaments. Parce que ce processus dépend de l’empaquetage de l’ADN, et non de mutations permanentes, il est potentiellement interrompable. Les résultats soutiennent des stratégies de traitement associant médicaments ciblés et agents bloquant la signalisation NF‑κB/RelA ou ses partenaires modifiant la chromatine, en particulier dans les tumeurs dont la chromatine est prédisposée à ce type de plasticité. En empêchant les cellules d’entrer en état persistant, de telles combinaisons pourraient rendre les rémissions plus profondes et plus durables.
Citation: Su, Y., Liu, C., Lu, X. et al. Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer. Nat Commun 17, 3228 (2026). https://doi.org/10.1038/s41467-026-71349-4
Mots-clés: cellules persistantes tolérantes aux médicaments, signalisation NF-kB RelA, remodelage chromatinaire, plasticité des cellules cancéreuses, résistance aux thérapies ciblées