Clear Sky Science · en
Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer
Why cancer cells can bounce back after treatment
Modern cancer drugs can shrink tumors dramatically, yet all too often the disease returns. This study explores a hidden escape route that cancer cells use: instead of being permanently killed or mutating, some cells temporarily “rewind” to a more primitive, stem‑like state that helps them survive therapy. By tracking these changes over time at the levels of genes and chromatin (the way DNA is packaged), the authors uncover a step‑by‑step program, controlled by a stress‑sensing pathway called NF‑κB/RelA, that lets cancer cells enter and later leave a drug‑tolerant state. Understanding this reversible process suggests new ways to keep treatments effective for longer.
Cells that survive by becoming less specialized
The researchers focused on melanomas driven by mutant BRAF, a common target of precision drugs. When they treated highly adaptable melanoma cells with a BRAF inhibitor, the cells did not simply die or stay the same. Instead they moved through a sequence of identities: from pigment‑producing melanoma cells, to a neural‑crest‑like state that divides slowly and tolerates drugs, and finally to an even more primitive, mesenchymal‑like state that is highly resistant to many therapies. Remarkably, when the drug was removed, these same cells gradually returned to their original, drug‑sensitive identity, showing that the resistance was not due to permanent DNA mutations but to a flexible change of state.
A looping path rather than a straight line
To make sense of thousands of changing genes, the team used an information‑theory–based method to compress the data into just two major “waves” of coordinated gene activity. The first, an early wave, turned on within days of treatment and was linked to rapid shifts in chromatin and a halt in cell division. The second, later wave controlled genes that define cell identity and invasive behavior. Plotting these two waves against each other revealed that the path into drug tolerance and the path back out did not overlap: cells remembered their treatment history in the configuration of their chromatin, a behavior known as hysteresis. This memory means that even when gene activity looks similar, the underlying DNA packaging can be very different.
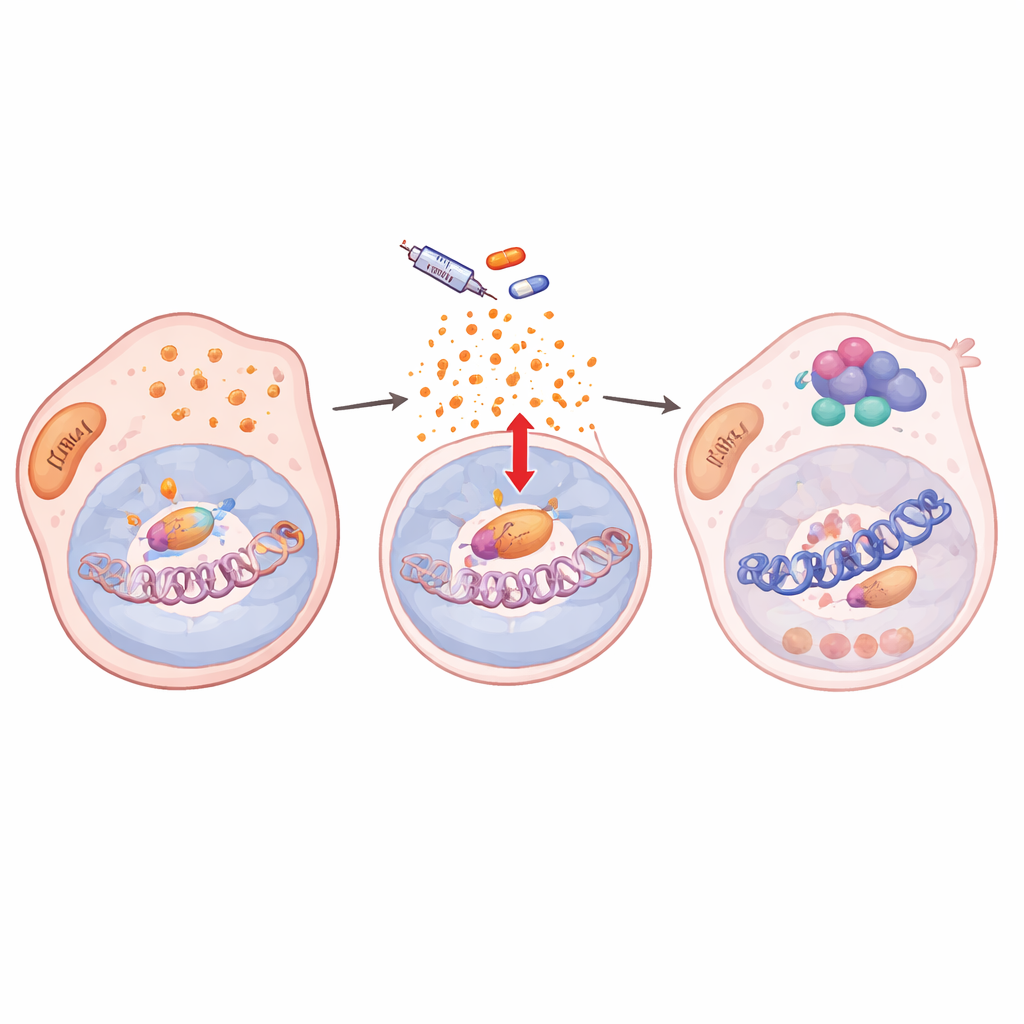
How stress signals rewire DNA packaging
Diving deeper, the authors found that drug treatment disturbed the cells’ antioxidant defenses, leading to a build‑up of reactive oxygen species (ROS). This oxidative stress activated NF‑κB/RelA, a master stress‑response factor. Once activated, RelA moved into the nucleus and bound many sites in the genome together with two chromatin‑modifying enzymes, KDM5B and HDAC1. At hundreds of target genes, including key melanoma identity genes such as SOX10 and MITF, this trio stripped away activating histone marks and made local chromatin less accessible. Gene activity dropped and stayed low as long as the drug was present, locking cells into a slow‑cycling, drug‑tolerant state. When RelA’s nuclear entry was blocked, chromatin marks were restored, SOX10 rebounded, and cells became sensitive to BRAF inhibition again.
Plasticity encoded in chromatin, shared across cancers
The team then asked why some tumors are more prone to this shape‑shifting behavior than others. Across several melanoma cell lines, they found that the degree of drug‑induced change tracked with how open and active the chromatin at RelA target genes was before treatment. Lines with more “permissive” chromatin showed larger transitions and stronger benefit from combining BRAF inhibitors with drugs targeting RelA or its chromatin partners. Finally, they showed that a similar ROS–NF‑κB–chromatin pathway drives entry into drug‑tolerant persister states in lung and colon cancers treated against their own driver oncogenes. In all cases, blocking NF‑κB/RelA activation or dampening ROS curtailed the emergence and regrowth of persister cells.
What this means for future cancer treatments
This work recasts drug resistance in many cancers as a reversible stress‑response program rather than solely a genetic problem. Cancer cells sense therapy‑induced oxidative stress, activate NF‑κB/RelA, and use associated chromatin remodelers to temporarily switch off genes that keep them specialized and drug‑sensitive. Because this process depends on how DNA is packaged, and not on permanent mutations, it can potentially be interrupted. The findings support treatment strategies that pair targeted drugs with agents that block NF‑κB/RelA signaling or its chromatin‑modifying partners, especially in tumors whose chromatin is primed for such plasticity. By preventing cells from entering the persister state in the first place, such combinations might make remissions deeper and more durable.
Citation: Su, Y., Liu, C., Lu, X. et al. Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer. Nat Commun 17, 3228 (2026). https://doi.org/10.1038/s41467-026-71349-4
Keywords: drug-tolerant persister cells, NF-kB RelA signaling, chromatin remodeling, cancer cell plasticity, targeted therapy resistance