Clear Sky Science · pl
Fale sekwencyjnej transkrypcji i przebudowa chromatyny napędzana przez NF-κB kierują indukowaną lekami dedyferencjacją w nowotworach
Dlaczego komórki nowotworowe potrafią odrodzić się po leczeniu
Nowoczesne leki przeciwnowotworowe potrafią znacząco zmniejszyć guzy, ale choroba zbyt często powraca. W badaniu zbadano ukryte drogi ucieczki, z których korzystają komórki nowotworowe: zamiast zostać trwale zabitymi lub ulec mutacjom, niektóre komórki tymczasowo „przewijają” się do bardziej prymitywnego, przypominającego komórki macierzyste stanu, co pomaga im przetrwać terapię. Śledząc te zmiany w czasie na poziomie genów i chromatyny (sposób upakowania DNA), autorzy odkrywają programu krok po kroku, kontrolowany przez odpowiadającą na stres ścieżkę NF-κB/RelA, który pozwala komórkom nowotworowym wejść i później opuścić stan tolerancji na lek. Zrozumienie tego odwracalnego procesu sugeruje nowe sposoby utrzymania skuteczności terapii przez dłuższy czas.
Komórki, które przetrwają, stając się mniej wyspecjalizowane
Naukowcy skupili się na czerniakach z napędem przez zmutowany BRAF, często cechowanych jako cel terapii precyzyjnej. Po leczeniu wysoce adaptowalnych komórek czerniaka inhibitorem BRAF, komórki nie po prostu umierały ani nie pozostawały bez zmian. Zamiast tego przechodziły przez sekwencję tożsamości: od melanocytów produkujących pigment, przez stan przypominający grzebień nerwowy, charakteryzujący się wolnym dzieleniem i tolerancją na leki, aż do jeszcze bardziej prymitywnego, podobnego do mezenchymy stanu, który jest wysoce oporny na wiele terapii. Co istotne, po usunięciu leku te same komórki stopniowo powracały do pierwotnej, wrażliwej na lek tożsamości, co pokazuje, że oporność nie wynikała z trwałych mutacji DNA, lecz z elastycznej zmiany stanu.
Ścieżka w pętli, a nie linia prosta
Aby zrozumieć tysiące zmieniających się genów, zespół zastosował metodę opartą na teorii informacji, która skompresowała dane do zaledwie dwóch głównych „fal” skoordynowanej aktywności genów. Pierwsza, wczesna fala, włączała się w ciągu dni od rozpoczęcia leczenia i była związana z szybkimi zmianami w chromatynie oraz zatrzymaniem podziałów komórkowych. Druga, późniejsza fala kontrolowała geny definiujące tożsamość komórkową i zachowania inwazyjne. Wykreślenie tych dwóch fal względem siebie ujawniło, że droga w kierunku tolerancji na lek i droga powrotu nie pokrywały się: komórki „pamiętały” historię leczenia w konfiguracji swojej chromatyny — zjawisko znane jako histereza. Ta pamięć oznacza, że nawet gdy aktywność genów wygląda podobnie, będące u jej podstaw upakowanie DNA może być bardzo różne.
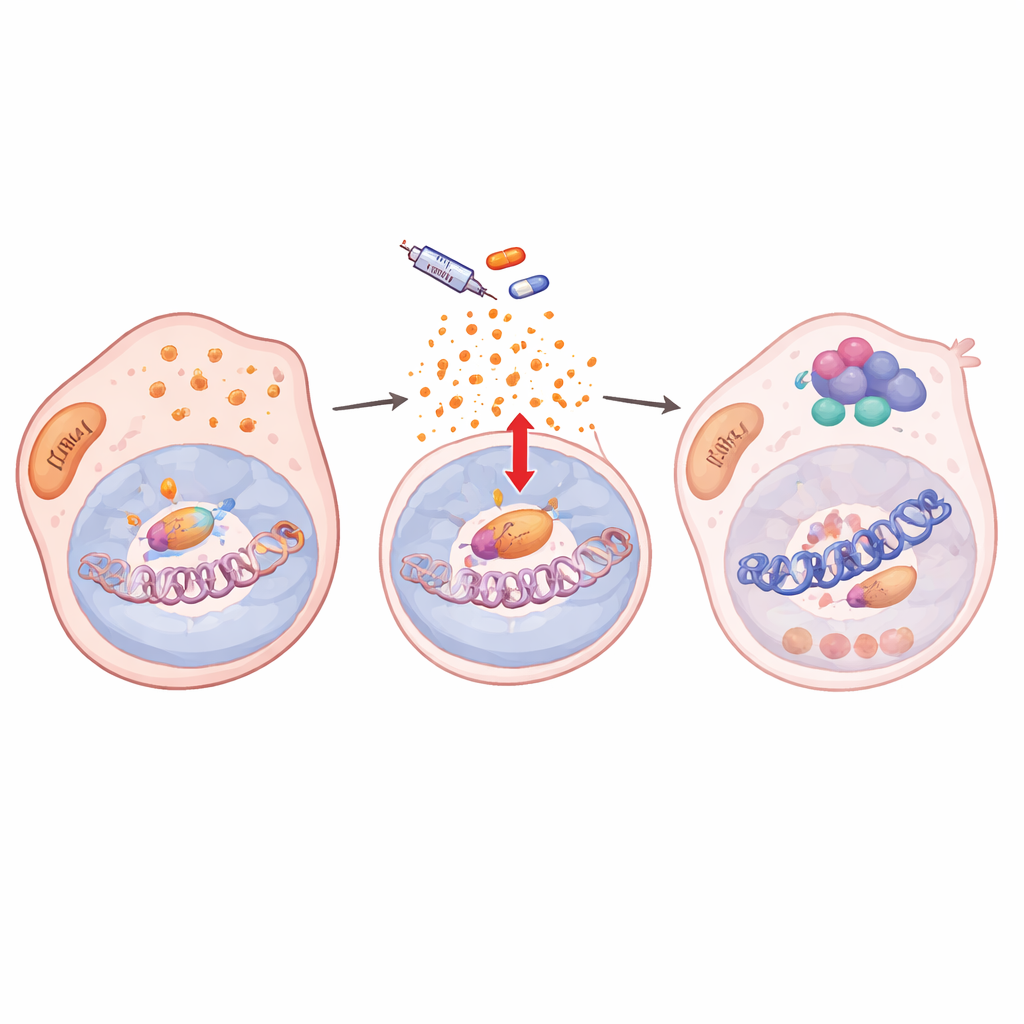
Jak sygnały stresowe przebudowują upakowanie DNA
Zagłębiając się dalej, autorzy stwierdzili, że leczenie zaburzało antyoksydacyjne mechanizmy obronne komórek, prowadząc do nagromadzenia reaktywnych form tlenu (ROS). Ten stres oksydacyjny aktywował NF-κB/RelA, główny czynnik odpowiedzi na stres. Po aktywacji RelA przemieszczał się do jądra i łączył na wielu miejscach genomu z dwoma enzymami modyfikującymi chromatynę, KDM5B i HDAC1. Przy setkach genów docelowych, w tym kluczowych genach tożsamości melanocytów takich jak SOX10 i MITF, to trio usuwało aktywujące modyfikacje histonów i zmniejszało lokalną dostępność chromatyny. Aktywność genów spadała i pozostawała niska tak długo, jak obecny był lek, blokując komórki w stanie wolnego cyklu i tolerancji na lek. Gdy zablokowano wejście RelA do jądra, markery chromatyny przywracały się, SOX10 wracał do normy, a komórki ponownie stawały się wrażliwe na hamowanie BRAF.
Plastyczność zakodowana w chromatynie, wspólna dla różnych nowotworów
Zespół zapytał następnie, dlaczego niektóre guzy są bardziej podatne na takie zmiany niż inne. W kilku liniach komórkowych czerniaka stwierdzono, że stopień zmiany indukowanej lekiem korelował z tym, jak otwarta i aktywna była chromatyna przy genach docelowych RelA przed leczeniem. Linie z bardziej „permissywną” chromatyną wykazywały większe przejścia i silniejszy efekt łączenia inhibitorów BRAF z lekami celującymi RelA lub jego partnerów chromatynowych. Wreszcie wykazano, że podobna ścieżka ROS–NF-κB–chromatyna napędza wejście w stan przetrwalnikowy tolerujący leki także w rakach płuca i jelita grubego leczonych przeciw ich własnym onkogenom napędowym. We wszystkich przypadkach blokowanie aktywacji NF-κB/RelA lub tłumienie ROS ograniczało pojawianie się i ponowny wzrost komórek przetrwalnikowych.
Co to oznacza dla przyszłych terapii nowotworowych
Ta praca przekształca postrzeganie oporności na leki w wielu nowotworach z wyłącznie problemu genetycznego w odwracalny program reakcji na stres. Komórki nowotworowe wykrywają indukowany terapią stres oksydacyjny, aktywują NF-κB/RelA i wykorzystują związane z nim remodelery chromatyny do tymczasowego wyłączania genów odpowiedzialnych za ich wyspecjalizowanie i wrażliwość na leki. Ponieważ proces ten zależy od upakowania DNA, a nie od trwałych mutacji, można go potencjalnie przerwać. Wyniki wspierają strategie terapeutyczne łączące leki celowane z czynnikami blokującymi sygnalizację NF-κB/RelA lub jego partnerów chromatynowych, szczególnie w guzach, których chromatyna jest przygotowana do takiej plastyczności. Zapobiegając wejściu komórek w stan przetrwalnikowy, takie kombinacje mogą uczynić remisje głębszymi i trwalszymi.
Cytowanie: Su, Y., Liu, C., Lu, X. et al. Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer. Nat Commun 17, 3228 (2026). https://doi.org/10.1038/s41467-026-71349-4
Słowa kluczowe: komórki przetrwalnikowe tolerujące leki, sygnalizacja NF-kB RelA, przebudowa chromatyny, plastyczność komórek nowotworowych, oporność na terapię celowaną