Clear Sky Science · pt
Ondas transcricionais sequenciais e remodelamento cromatínico dirigido por NF-κB orientam a desdiferenciação induzida por drogas no câncer
Por que células cancerosas podem se recuperar após o tratamento
Medicamentos oncológicos modernos podem reduzir tumores de forma dramática, mas, com muita frequência, a doença retorna. Este estudo investiga uma rota de escape oculta que as células cancerosas usam: em vez de serem permanentemente eliminadas ou de acumularem mutações, algumas células “retrocedem” temporariamente a um estado mais primitivo, semelhante a células-tronco, que as ajuda a sobreviver à terapia. Ao acompanhar essas mudanças ao longo do tempo nos níveis de genes e cromatina (a forma como o DNA é empacotado), os autores revelam um programa passo a passo, controlado por uma via sensora de estresse chamada NF-κB/RelA, que permite que as células cancerosas entrem e posteriormente saiam de um estado tolerante ao fármaco. Compreender esse processo reversível sugere novas maneiras de manter os tratamentos eficazes por mais tempo.
Células que sobrevivem tornando-se menos especializadas
Os pesquisadores concentraram-se em melanomas impulsionados por BRAF mutante, um alvo comum de medicamentos de precisão. Quando trataram células de melanoma altamente adaptáveis com um inibidor de BRAF, as células não simplesmente morreram nem permaneceram inalteradas. Em vez disso, elas percorreram uma sequência de identidades: de células produtoras de pigmento, para um estado semelhante à crista neural que se divide lentamente e tolera drogas, e finalmente para um estado ainda mais primitivo, semelhante ao mesenquimal, altamente resistente a muitas terapias. Notavelmente, quando a droga foi retirada, essas mesmas células gradualmente retornaram à sua identidade original, sensível ao fármaco, mostrando que a resistência não se devia a mutações permanentes no DNA, mas a uma mudança de estado flexível.
Um caminho em loop em vez de uma linha reta
Para interpretar milhares de genes em mudança, a equipe usou um método baseado em teoria da informação para comprimir os dados em apenas duas grandes “ondas” coordenadas de atividade gênica. A primeira, uma onda precoce, se ativou dentro de dias após o tratamento e esteve ligada a mudanças rápidas na cromatina e a uma pausa na divisão celular. A segunda, posterior, controlou genes que definem identidade celular e comportamento invasivo. Traçar essas duas ondas uma contra a outra revelou que o caminho de entrada na tolerância à droga e o caminho de saída não se sobrepunham: as células lembravam seu histórico de tratamento na configuração de sua cromatina, um comportamento conhecido como histerese. Essa memória significa que, mesmo quando a atividade gênica parece similar, o empacotamento subjacente do DNA pode ser muito diferente.
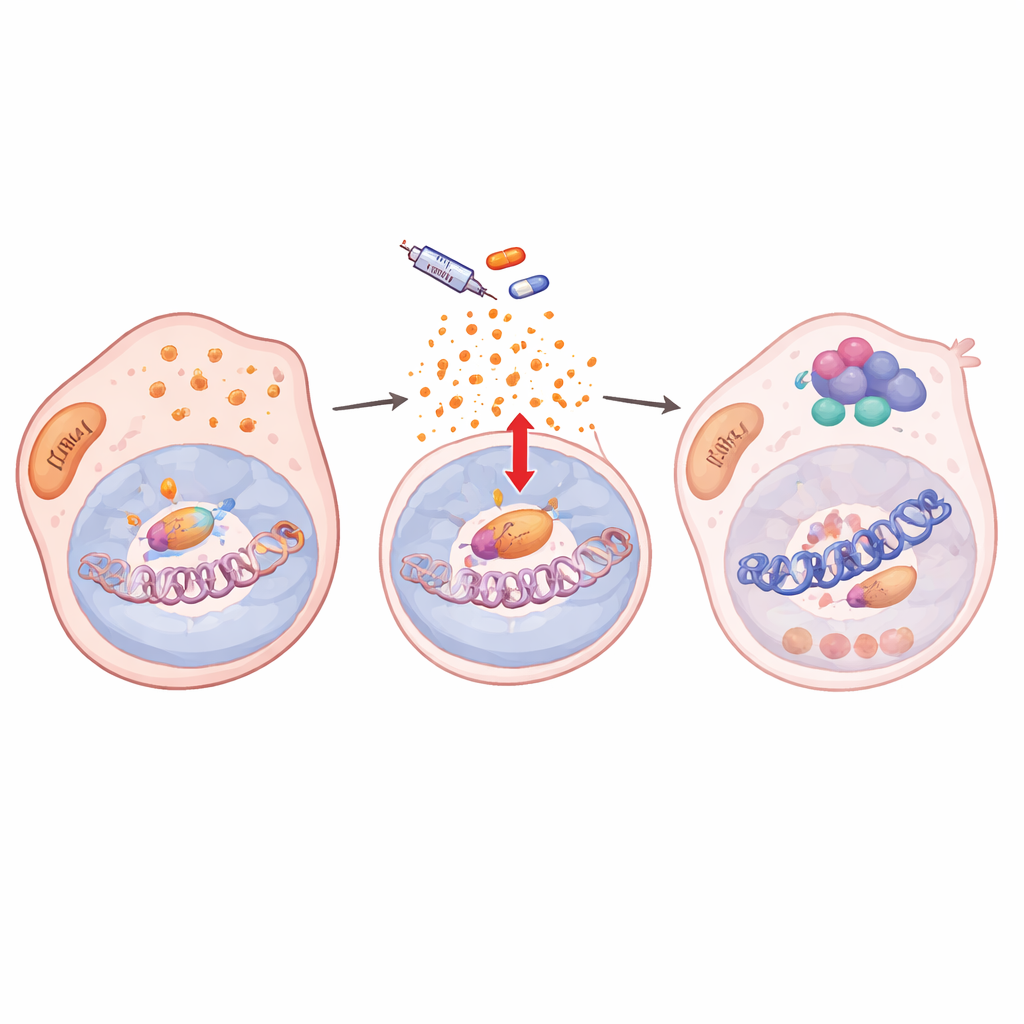
Como sinais de estresse reprogramam o empacotamento do DNA
Ao aprofundar, os autores descobriram que o tratamento com a droga perturbou as defesas antioxidantes das células, levando ao acúmulo de espécies reativas de oxigênio (ROS). Esse estresse oxidativo ativou NF-κB/RelA, um fator mestre da resposta ao estresse. Uma vez ativado, RelA deslocou-se para o núcleo e se ligou a muitos locais do genoma juntamente com duas enzimas modificadoras de cromatina, KDM5B e HDAC1. Em centenas de genes-alvo, incluindo genes-chave de identidade do melanoma como SOX10 e MITF, esse trio removeu marcas historiônicas ativadoras e tornou a cromatina local menos acessível. A atividade gênica caiu e permaneceu baixa enquanto a droga estava presente, prendendo as células em um estado de ciclo lento e tolerante ao fármaco. Quando a entrada nuclear de RelA foi bloqueada, as marcas na cromatina foram restauradas, SOX10 se recuperou e as células novamente se tornaram sensíveis à inibição de BRAF.
Plasticidade codificada na cromatina, compartilhada entre cânceres
Em seguida, a equipe investigou por que alguns tumores são mais propensos a esse comportamento de mudança de forma do que outros. Em várias linhas celulares de melanoma, eles identificaram que o grau de mudança induzida pela droga acompanhava o quão aberta e ativa a cromatina nos genes-alvo de RelA estava antes do tratamento. Linhas com cromatina mais “permissiva” mostraram transições maiores e um benefício mais forte ao combinar inibidores de BRAF com drogas que visam RelA ou seus parceiros de cromatina. Finalmente, demonstraram que uma via semelhante ROS–NF-κB–cromatina impulsiona a entrada em estados persistentes tolerantes a drogas em cânceres de pulmão e de cólon tratados contra seus próprios oncogenes condutores. Em todos os casos, bloquear a ativação de NF-κB/RelA ou atenuar o ROS reduziu o surgimento e o re crescimento das células persistentes.
O que isso significa para tratamentos oncológicos futuros
Este trabalho reconceptualiza a resistência a drogas em muitos cânceres como um programa reversível de resposta ao estresse, em vez de apenas um problema genético. As células cancerosas percebem o estresse oxidativo induzido pela terapia, ativam NF-κB/RelA e usam remodeladores de cromatina associados para desligar temporariamente genes que as mantêm especializadas e sensíveis aos fármacos. Como esse processo depende de como o DNA é empacotado, e não de mutações permanentes, ele pode potencialmente ser interrompido. As descobertas apoiam estratégias terapêuticas que combinam fármacos alvo com agentes que bloqueiam a sinalização NF-κB/RelA ou seus parceiros modificadores de cromatina, especialmente em tumores cuja cromatina está preparada para tal plasticidade. Ao impedir que as células entrem no estado persistente desde o início, tais combinações podem tornar as remissões mais profundas e duradouras.
Citação: Su, Y., Liu, C., Lu, X. et al. Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer. Nat Commun 17, 3228 (2026). https://doi.org/10.1038/s41467-026-71349-4
Palavras-chave: células persistentes tolerantes a drogas, sinalização NF-kB RelA, remodelamento cromatínico, plasticidade de células cancerosas, resistência à terapia alvo