Clear Sky Science · es
Olas transcripcionales secuenciales y remodelado de la cromatina dirigido por NF-κB guían la desdiferenciación inducida por fármacos en el cáncer
Por qué las células cancerosas pueden recuperarse tras el tratamiento
Los fármacos oncológicos modernos pueden reducir tumores de forma notable, sin embargo con demasiada frecuencia la enfermedad reaparece. Este estudio explora una vía de escape oculta que usan las células cancerosas: en lugar de ser eliminadas de forma permanente o mutar, algunas células «rebobinan» temporalmente a un estado más primitivo, similar al de células madre, que les ayuda a sobrevivir la terapia. Al seguir estos cambios a lo largo del tiempo en los niveles de genes y de la cromatina (la forma en que se empaqueta el ADN), los autores descubren un programa paso a paso, controlado por una vía sensorade estrés llamada NF‑κB/RelA, que permite a las células cancerosas entrar y luego salir de un estado tolerante a fármacos. Comprender este proceso reversible sugiere nuevas formas de mantener la eficacia de los tratamientos durante más tiempo.
Células que sobreviven volviéndose menos especializadas
Los investigadores se centraron en melanomas impulsados por la mutación BRAF, un objetivo común de los fármacos de precisión. Cuando trataron células de melanoma altamente adaptables con un inhibidor de BRAF, las células no se limitaron a morir o a mantenerse igual. En su lugar, atravesaron una secuencia de identidades: desde células de melanoma productoras de pigmento, a un estado similar a la cresta neural que se divide despacio y tolera los fármacos, y finalmente a un estado aún más primitivo, similar al mesenquimal, que es altamente resistente a muchas terapias. De manera notable, cuando se retiró el fármaco, esas mismas células regresaron gradualmente a su identidad original sensible al fármaco, lo que muestra que la resistencia no se debía a mutaciones permanentes del ADN sino a un cambio de estado flexible.
Un camino en bucle en lugar de una línea recta
Para entender miles de genes cambiantes, el equipo utilizó un método basado en la teoría de la información para comprimir los datos en solo dos «olas» principales de actividad génica coordinada. La primera, una ola temprana, se activó en días tras el tratamiento y se vinculó a cambios rápidos en la cromatina y a una detención de la división celular. La segunda, más tardía, controló genes que definen la identidad celular y el comportamiento invasivo. Al representar estas dos olas entre sí se reveló que la ruta hacia la tolerancia al fármaco y la ruta de regreso no se solapaban: las células recordaban su historia de tratamiento en la configuración de su cromatina, un comportamiento conocido como histeresis. Esta memoria implica que, aun cuando la actividad génica parezca similar, el empaquetamiento subyacente del ADN puede ser muy distinto.
Cómo las señales de estrés reconfiguran el empaquetamiento del ADN
Al profundizar, los autores hallaron que el tratamiento con fármacos alteraba las defensas antioxidantes de las células, provocando una acumulación de especies reactivas de oxígeno (ROS). Este estrés oxidativo activó NF‑κB/RelA, un regulador maestro de la respuesta al estrés. Una vez activado, RelA se desplazó al núcleo y se unió en muchos sitios del genoma junto con dos enzimas modificadoras de la cromatina, KDM5B y HDAC1. En cientos de genes diana, incluidos genes clave de identidad del melanoma como SOX10 y MITF, este trío eliminó marcas de histonas activadoras e hizo la cromatina local menos accesible. La actividad génica se redujo y permaneció baja mientras el fármaco estuvo presente, bloqueando a las células en un estado de ciclo lento y tolerante al fármaco. Cuando se impidió la entrada nuclear de RelA, las marcas de cromatina se restauraron, SOX10 se recuperó y las células volvieron a ser sensibles a la inhibición de BRAF.
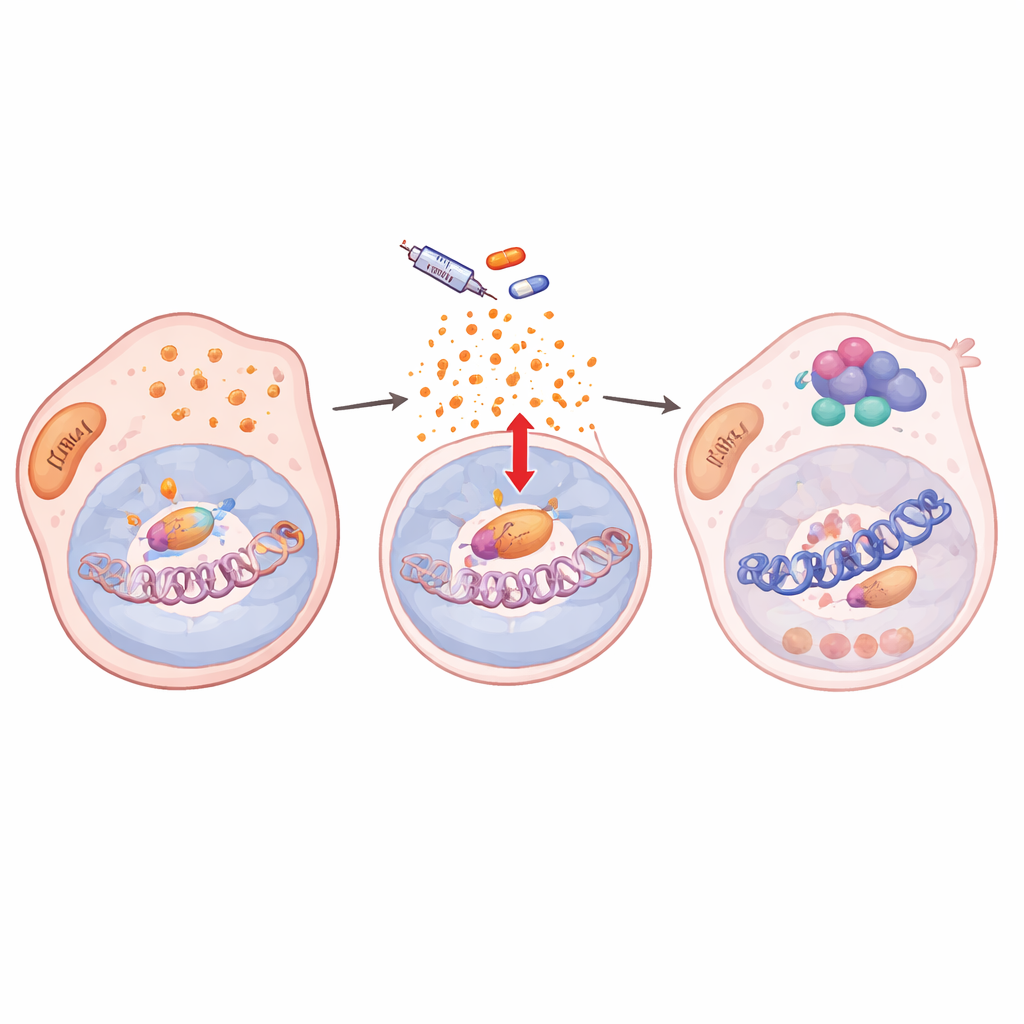
Plasticidad codificada en la cromatina, compartida entre distintos cánceres
El equipo preguntó entonces por qué algunos tumores son más propensos a este cambio de forma que otros. En varias líneas celulares de melanoma, encontraron que el grado de cambio inducido por el fármaco se correspondía con cuán abierta y activa estaba la cromatina en los genes diana de RelA antes del tratamiento. Las líneas con una cromatina más «permisiva» mostraron transiciones mayores y un beneficio más fuerte al combinar inhibidores de BRAF con fármacos que atacan a RelA o sus socios en la cromatina. Finalmente, demostraron que una vía similar ROS–NF‑κB–cromatina impulsa la entrada en estados persistentes tolerantes a fármacos en cánceres de pulmón y colon tratados contra sus propios oncogenes conductores. En todos los casos, bloquear la activación de NF‑κB/RelA o reducir las ROS limitó la aparición y el re‑crecimiento de las células persistentes.
Qué supone esto para los tratamientos futuros contra el cáncer
Este trabajo replantea la resistencia a fármacos en muchos cánceres como un programa reversible de respuesta al estrés y no únicamente como un problema genético. Las células cancerosas detectan el estrés oxidativo inducido por la terapia, activan NF‑κB/RelA y emplean remodeladores de la cromatina asociados para apagar temporalmente genes que las mantienen especializadas y sensibles al fármaco. Debido a que este proceso depende de cómo está empaquetado el ADN, y no de mutaciones permanentes, puede potencialmente interrumpirse. Los hallazgos respaldan estrategias terapéuticas que combinen fármacos dirigidos con agentes que bloqueen la señalización de NF‑κB/RelA o sus socios modificadores de la cromatina, especialmente en tumores cuya cromatina está preparada para tal plasticidad. Al impedir que las células entren en el estado persistente desde el principio, tales combinaciones podrían hacer que las remisiones sean más profundas y duraderas.
Cita: Su, Y., Liu, C., Lu, X. et al. Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer. Nat Commun 17, 3228 (2026). https://doi.org/10.1038/s41467-026-71349-4
Palabras clave: células persistentes tolerantes a fármacos, señalización NF-kB RelA, remodelado de la cromatina, plasticidad de las células cancerosas, resistencia a terapias dirigidas