Clear Sky Science · sv
Sekventiella transkriptionsvågor och NF-κB-styrd kromatinombyggnad styr läkemedelsinducerad dedifferentiering i cancer
Varför cancerceller kan studsa tillbaka efter behandling
Moderna cancerläkemedel kan krympa tumörer dramatiskt, men alltför ofta återkommer sjukdomen. Denna studie utforskar en dold flyktväg som cancerceller använder: i stället för att bli permanent dödade eller mutera, går vissa celler tillfälligt tillbaka till ett mer primitivt, stamliknande tillstånd som hjälper dem att överleva behandlingen. Genom att följa dessa förändringar över tid på nivåerna av gener och kromatin (hur DNA är paketerat) identifierar författarna ett steg‑för‑steg‑program, styrt av en stresskänslig bana kallad NF‑κB/RelA, som låter cancerceller gå in i och senare lämna ett läkemedelstolerant tillstånd. Att förstå denna reversibla process pekar ut nya sätt att hålla behandlingar effektiva längre.
Celler som överlever genom att bli mindre specialiserade
Forskarna fokuserade på melanom drivna av muterat BRAF, ett vanligt mål för precisionsläkemedel. När de behandlade mycket anpassningsbara melanomceller med en BRAF‑hämmare dog cellerna inte eller förblev oförändrade. I stället gick de igenom en sekvens av identiteter: från pigmentproducerande melanomceller till ett neural‑crest‑liknande tillstånd som delar sig långsamt och tolererar läkemedel, och slutligen till ett ännu mer primitivt, mesenkym‑likt tillstånd som är starkt resistent mot många terapier. Märkbart var att när läkemedlet avlägsnades återvände dessa samma celler gradvis till sin ursprungliga, läkemedelskänsliga identitet, vilket visar att resistensen inte berodde på permanenta DNA‑mutationer utan på en flexibel tillståndsförändring.
En loopande väg snarare än en rak linje
För att förstå tusentals föränderliga gener använde teamet en informations‑teoribaserad metod för att komprimera data till bara två stora ”vågor” av koordinerad genaktivitet. Den första, en tidig våg, aktiverades inom några dagar efter behandling och kopplades till snabba skift i kromatin och ett stopp i celldelningen. Den andra, senare vågen styrde gener som definierar cellidentitet och invasivt beteende. Genom att plottas mot varandra visade dessa två vågor att vägen in i läkemedelstolerans och vägen ut inte överlappade: cellerna bar minnet av sin behandling i konfigurationen av sitt kromatin, ett beteende känt som hysteresis. Detta minne innebär att även när genaktiviteten ser liknande ut kan den underliggande DNA‑packningen vara mycket olika.
Hur stressignaler omkopplar DNA‑packningen
När författarna grävde djupare fann de att läkemedelsbehandlingen störde cellernas antioxidanter, vilket ledde till en uppbyggnad av reaktiva syreradikaler (ROS). Denna oxidativa stress aktiverade NF‑κB/RelA, en huvudregulator för stressrespons. När RelA aktiverades flyttade det in i kärnan och band många platser i genomet tillsammans med två kromatinmodifierande enzymer, KDM5B och HDAC1. Vid hundratals målade gener, inklusive viktiga melanomidentitetsgener såsom SOX10 och MITF, togs aktiverande histonmarkörer bort och det lokala kromatinet blev mindre tillgängligt. Genaktiviteten sjönk och förblev låg så länge läkemedlet var närvarande, vilket låste cellerna i ett långsamt delande, läkemedelstolerant tillstånd. När RelA:s kärntransport blockerades återställdes kromatinmarkörerna, SOX10 återhämtade sig och cellerna blev känsliga för BRAF‑hämning igen.
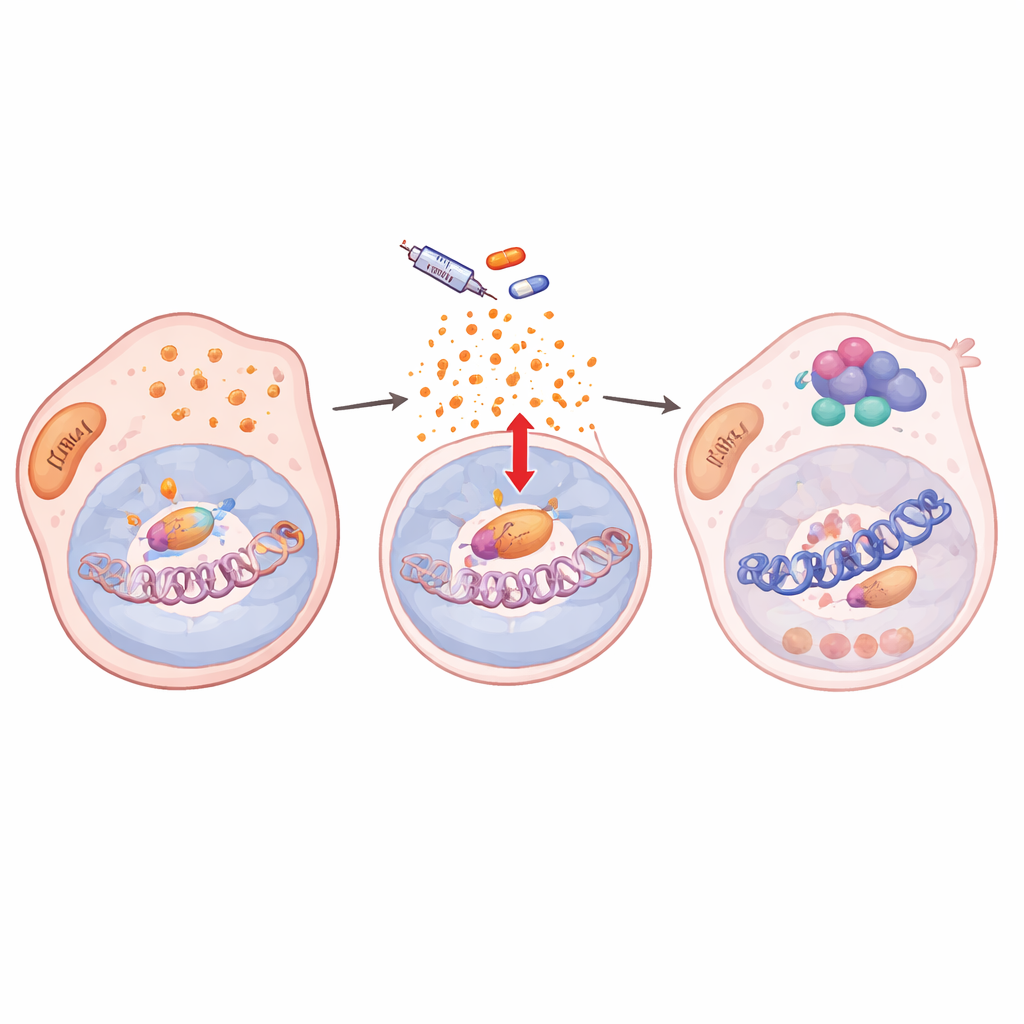
Plasticitet kodad i kromatin, delad över cancerformer
Teamet undrade sedan varför vissa tumörer är mer benägna till detta formskiftande beteende än andra. I flera melanomcellinjer fann de att graden av läkemedelsinducerad förändring korrelerade med hur öppet och aktivt kromatinet vid RelA‑målgener var före behandling. Linjer med mer ”tillåtande” kromatin visade större övergångar och starkare nytta av att kombinera BRAF‑hämmare med läkemedel som riktar sig mot RelA eller dess kromatinpartners. Slutligen visade de att en liknande ROS–NF‑κB–kromatin‑bana driver inträde i läkemedelstoleranta persistertillstånd i lung‑ och koloncancer som behandlas mot sina egna drivande onkogener. I samtliga fall dämpade blockering av NF‑κB/RelA‑aktivering eller minskning av ROS framväxten och återväxten av persisterceller.
Vad detta betyder för framtida cancerbehandlingar
Detta arbete omformulerar läkemedelsresistens i många cancerformer som ett reversibelt stressresponssprogram snarare än enbart ett genetiskt problem. Cancerceller känner av terapiinducerad oxidativ stress, aktiverar NF‑κB/RelA och använder associerade kromatinombyggare för att tillfälligt stänga av gener som håller dem specialiserade och läkemedelskänsliga. Eftersom denna process beror på hur DNA är paketerat, och inte på permanenta mutationer, kan den potentiellt avbrytas. Resultaten stöder behandlingsstrategier som parar ihop riktade läkemedel med ämnen som blockerar NF‑κB/RelA‑signalering eller dess kromatinmodifierande partners, särskilt i tumörer vars kromatin är förberett för sådan plasticitet. Genom att förhindra att celler går in i persistertillståndet från början kan sådana kombinationer göra remissioner djupare och mer bestående.
Citering: Su, Y., Liu, C., Lu, X. et al. Sequential transcriptional waves and NF-κB-driven chromatin remodeling direct drug-induced dedifferentiation in cancer. Nat Commun 17, 3228 (2026). https://doi.org/10.1038/s41467-026-71349-4
Nyckelord: läkemedels‑toleranta persisterceller, NF-kB RelA‑signalering, kromatinombyggnad, cancercellsplasticitet, motstånd mot riktad terapi