Clear Sky Science · zh
通过线性张量化四边形注意力构建可扩展且具量子精度的生物分子力场基础模型
为什么更快的分子“电影”很重要
现代药物发现和材料设计越来越依赖计算机生成的“电影”,来展示分子如何扭动、弯曲和反应。这些模拟可以揭示药物如何嵌入蛋白质口袋,或新材料在受力下的行为。但现有工具迫使科研人员做出权衡:要么选择快速但在精度上妥协的方法,要么采用极其精确但对于现实复杂体系过于缓慢的量子计算。本文介绍了 LiTEN 及其力场模型 LiTEN-FF,一种新的人工智能方法,旨在以适合日常分子建模的速度提供量子级别的精度。
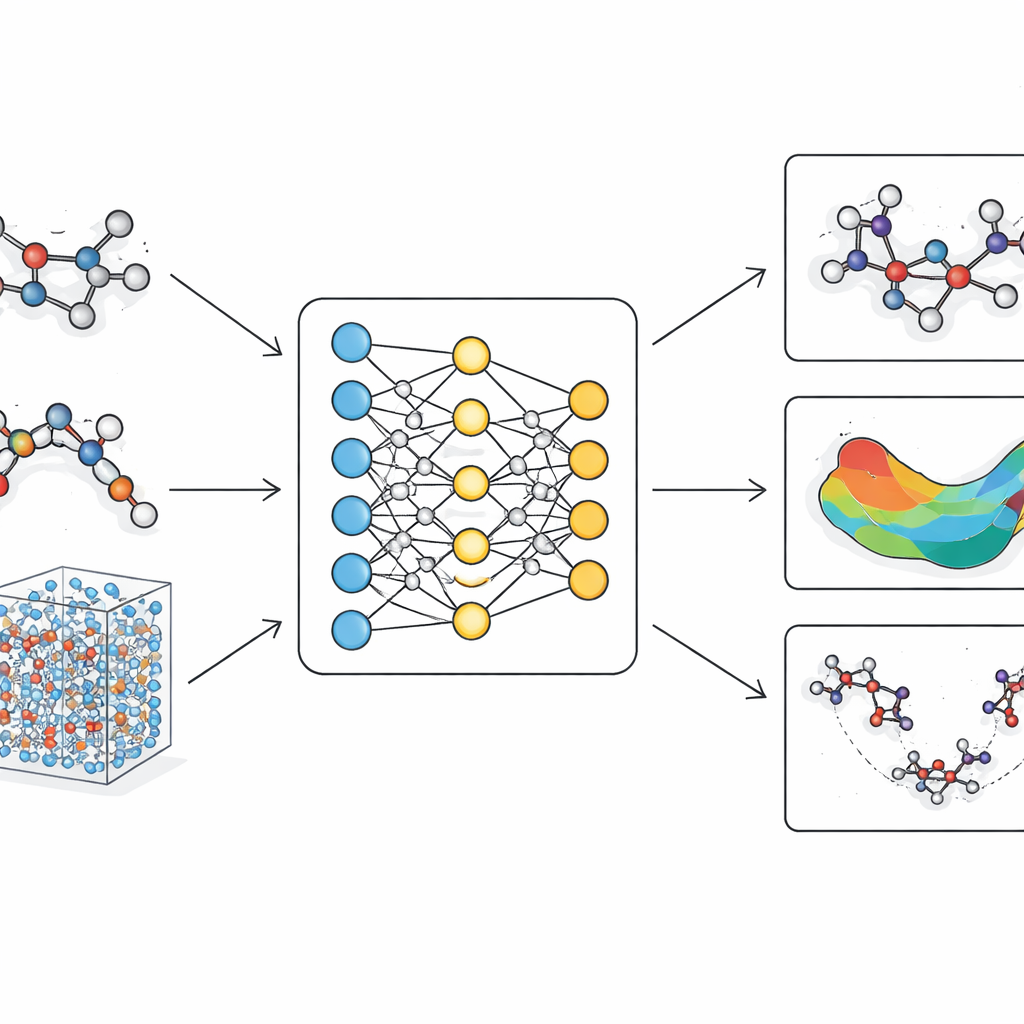
当今分子模型的问题
传统分子模拟大致分为两类。经典力场将原子视为由带固定参数的弹簧连接的粒子。它们运行迅速,能够处理大型蛋白或较长的时间尺度,但在处理细微构型变化、键重排和真实化学中关键的反应路径时显得力不从心。相比之下,量子方法显式描述电子,能够准确捕捉键的断裂与形成。然而,它们的计算需求极高,通常仅限于小分子或非常短的模拟。在过去几年中,机器学习方法作为中间道路出现,学习去模拟量子计算。但许多此类模型要么缺乏保证可靠性的物理严谨性,要么在扩展到大型、真实生物分子时变得过于缓慢。
一种教授 AI 关于分子形状的新方法
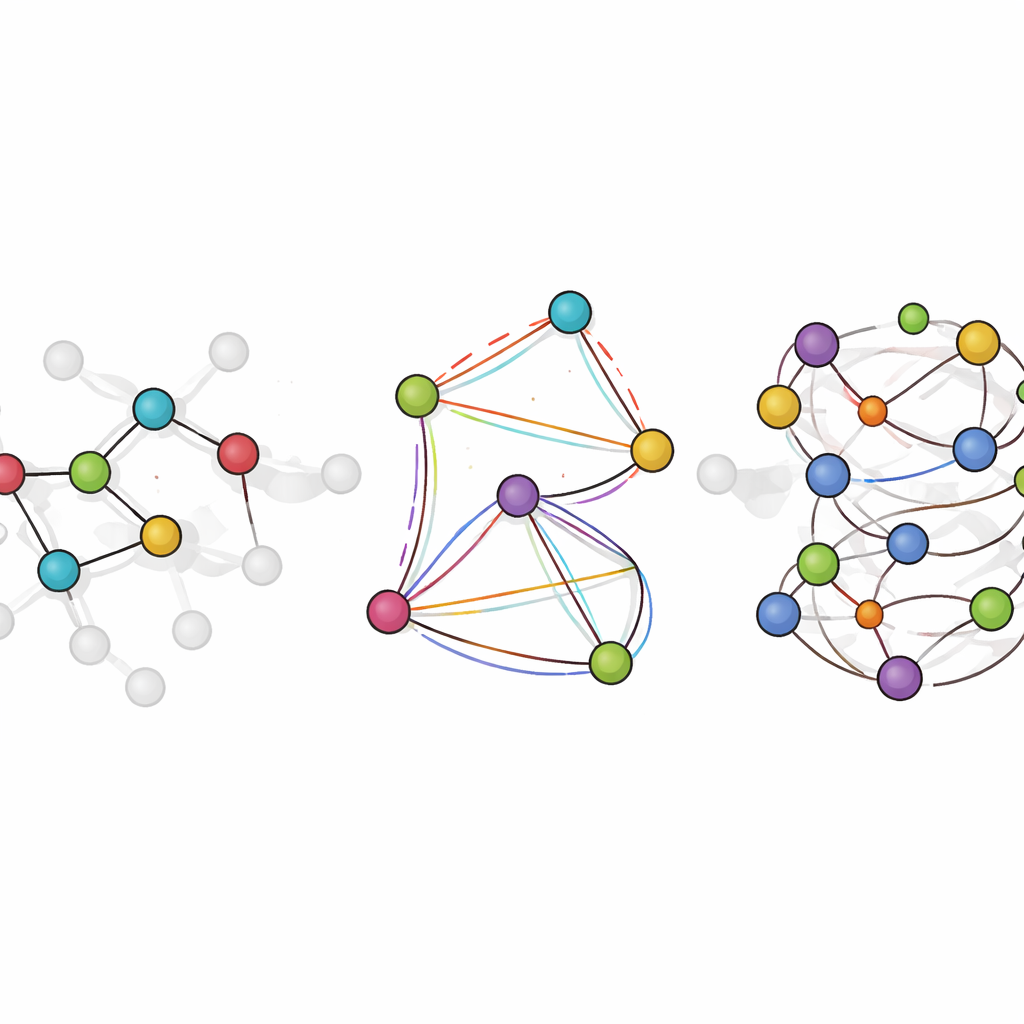
LiTEN 通过重新设计神经网络“感知”分子几何的方式来应对这一挑战。它不仅考虑原子之间的简单两体距离,还内建了关于控制分子中角度和扭转的三体与四体模式信息。关键在于,这一做法尊重基本物理对称性:若将分子在空间中旋转或移动,其预测能量保持不变,预测力则随之旋转。核心创新称为张量化四边形注意力,它使模型能够通过高效的向量运算而非繁重的专门数学,捕捉复杂的弯曲与扭转相互作用。这保持了计算的可扩展性,从而使通常会拖慢先进模型的多体效应以仅随体系规模线性增长的代价被处理。
从量子数据训练到真实生物分子
在此架构之上,作者构建了 LiTEN-FF,这是一种用于分子力学的“基础模型”。他们首先在包含数百万药物样分子的海量量子化学数据集上进行训练,然后在一个更小但精度更高的集合上细化,后者包括肽、溶剂化结构以及卤素和金属等多样元素。这个两阶段训练使模型既学习到广泛的化学覆盖面,又掌握精细细节。在将预测的能量与力与高阶量子结果比较的标准基准测试中,LiTEN 在小型刚性分子和更大、更具生物学相关性的体系上匹配或超越了领先模型。随着原子数增加到数百,它仍保持精度,同时比许多流行替代方案使用更少内存并运行更快。
在模拟化学中应用该模型
除了静态测试外,团队还在模拟真实研究工作流程的任务中评估了 LiTEN-FF。对于类药分子,它可以优化构型,使所得结构几乎与耗时的量子计算结果完全一致,但速度快了数千倍。在分子动力学运行中,它能重现键长和角度随时间的波动,紧密追踪基于量子的模拟。在预测分子绕关键键扭转时能量变化方面也表现出色,这对把握药物设计中的构象偏好至关重要。在液态水和短肽溶于水的体系中,LiTEN-FF 产生的结构和热力学性质与实验和更昂贵的参照模型均高度一致,同时在大型体系上提供高达十倍的加速。
加速有用构型的搜索
作者还展示了一个围绕 LiTEN-FF 构建的实用构象体搜索流程。通过反复进行高温动力学、冷却与快速几何精化循环,模型为复杂药物分子生成了丰富且各异的低能量构型集合。与一种广泛使用的基于量子的方法相比,这种由 AI 驱动的方法在不到一半的时间内找到了更多多样的构象。此外,由于 LiTEN-FF 能够并行处理许多分子而成本并不会成比例上升,它在需要评估数千个候选分子的规模化筛选活动中尤其强大。
这对未来药物与材料设计意味着什么
从本质上讲,LiTEN-FF 提供了一种新的分子模拟引擎,使量子级可靠性更接近日常可用。通过将角度与扭转的几何信息直接编码进高效的神经网络,它缩小了快速但近似的力场与缓慢但精确的量子计算之间的差距。对于非专业读者而言,结论是研究人员很快可能能够在计算机上进行更为现实的分子“实验”,达到与现代药物发现和材料开发相适应的规模与速度。如果被广泛采用并进一步优化,这一系列模型有望成为自动化流水线的核心组件,在分子被合成到实验室之前提出、测试并改进新分子。
引用: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
关键词: 分子模拟, 机器学习力场, 药物发现, 生物分子建模, 量子化学