Clear Sky Science · sv
En skalbar och kvantkemsikt korrekt grundmodell för biomolekylära kraftfält via linjärt tensoriserad fyrhörningsattention
Varför snabbare molekylfilmer spelar roll
Modern läkemedelsutveckling och materialdesign förlitar sig i allt högre grad på datorbaserade ”filmer” av molekyler som vrider sig, böjer sig och reagerar. Sådana simuleringar kan visa hur ett läkemedel lägger sig i ett proteinfack eller hur ett nytt material beter sig under påfrestning. Men dagens verktyg tvingar forskare att välja: snabba metoder som gör avkall på noggrannhet, eller extremt precisa kvantberäkningar som är alldeles för långsamma för realistiskt komplexa system. Denna artikel introducerar LiTEN och dess kraftfältsmodell LiTEN-FF, en ny artificiell intelligensmetod som syftar till att leverera kvantnivånoggrannhet i hastigheter lämpliga för vardaglig molekylmodellering.
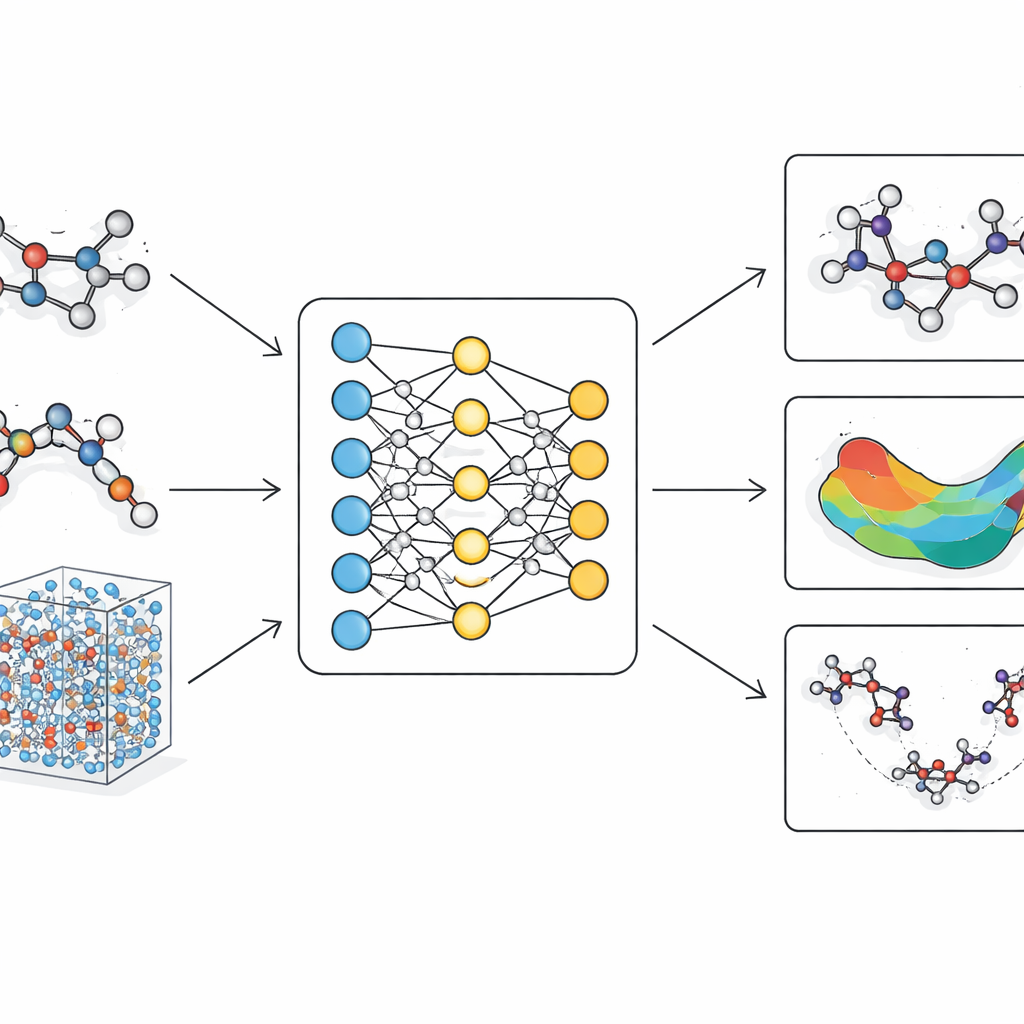
Problemet med dagens molekylmodeller
Traditionella molekylsimuleringar faller i två läger. Klassiska kraftfält behandlar atomer som kulor kopplade med fjädrar med fasta parametrar. De är snabba och kan hantera stora proteiner eller långa tidsskalor, men har svårt med subtila formändringar, bindningsomläggningar och reaktionsvägar som är viktiga för verklig kemi. Kvantmetoder beskriver däremot elektronerna explicit och kan fånga bindningsbrytning och -bildning på ett korrekt sätt. De är dock så beräkningsintensiva att de vanligtvis begränsas till små molekyler eller mycket korta simuleringar. Under de senaste åren har maskininlärningsmetoder dykt upp som en mellangata och lärt sig efterlikna kvantberäkningar. Många av dessa modeller saknar emellertid antingen den fysiska stringens som krävs för tillförlitlighet eller blir för långsamma när de skalas till stora, realistiska biomolekyler.
Ett nytt sätt att lära AI om molekylära former
LiTEN angriper denna utmaning genom att omforma hur ett neuralt nätverk ”känner” molekylgeometrin. I stället för att endast beakta enkla parvisa avstånd mellan atomer bygger LiTEN in information om tre- och fyra-atomsmönster som styr vinklar och vridningar i en molekyl. Viktigt är att detta görs samtidigt som grundläggande fysikaliska symmetrier respekteras: om du roterar eller förflyttar en molekyl i rummet förblir dess förutsagda energi densamma och de förutsagda krafterna roterar på ett korrekt sätt. Den centrala innovationen, kallad tensoriserad fyrhörningsattention, låter modellen fånga komplexa böjnings- och vridningsinteraktioner med effektiva vektoroperationer i stället för tung, specialiserad matematik. Det håller beräkningen skalbar, så många-kropps-effekter som vanligtvis sinkar avancerade modeller kan hanteras med en kostnad som bara växer linjärt med systemstorleken.
Från träning på kvantdata till riktiga biomolekyler
Ovanpå denna arkitektur bygger författarna LiTEN-FF, en ”grundmodell” för molekylära krafter. De tränar den först på en massiv kvantkemidatamängd med miljontals läkemedelsliknande molekyler och förfinar den därefter på en mindre men högre noggrannhetssamling som inkluderar peptider, solvatiserade strukturer och mångfald av element såsom halogener och metaller. Denna tvåstegsutbildning låter modellen lära både bred kemisk täckning och fin detaljkunskap. I standardbenchmarks som jämför förutsagda energier och krafter med högklassiga kvantresultat matchar eller överträffar LiTEN ledande modeller för både små, styva molekyler och mycket större, biologiskt relevanta system. Den behåller noggrannheten även när antalet atomer växer till hundratals, samtidigt som den använder mindre minne och kör snabbare än många populära alternativ.
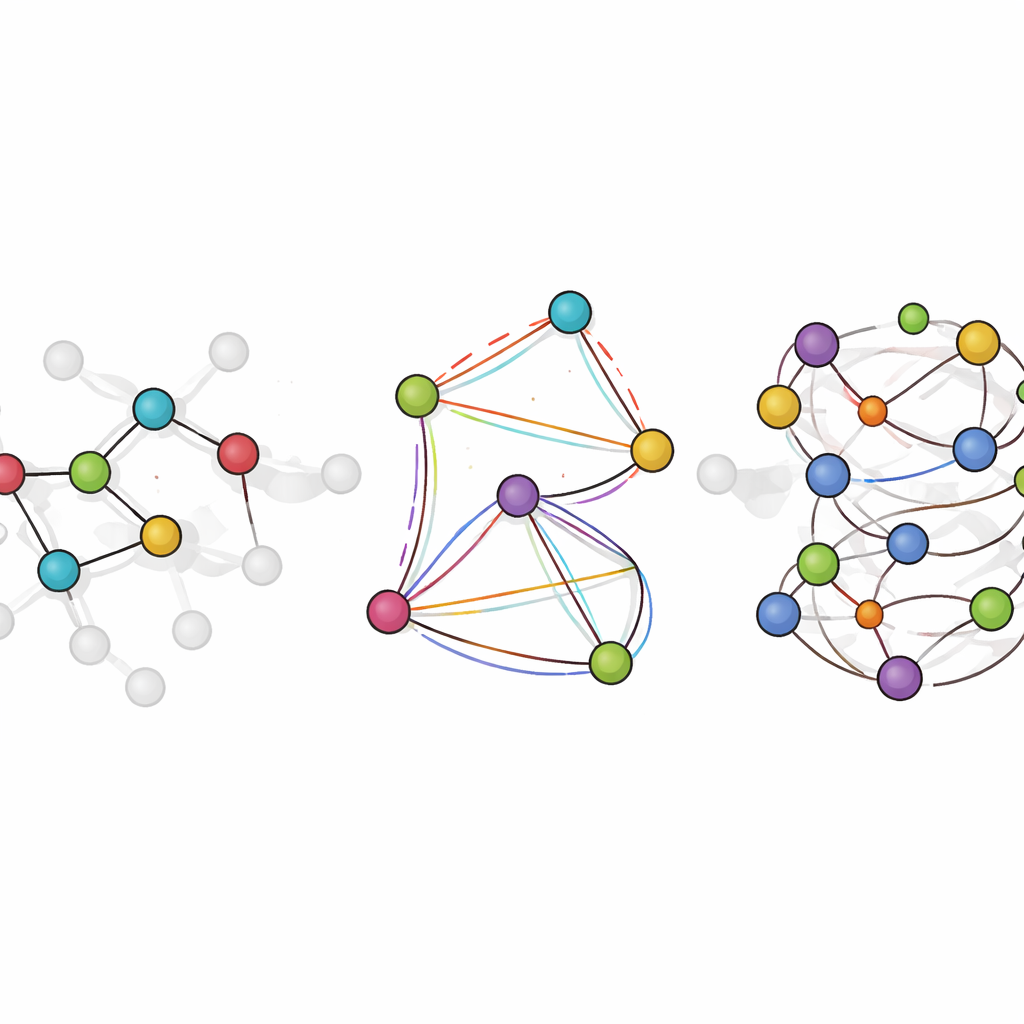
Användning av modellen i simulerad kemi
Bortom statiska tester utvärderar teamet LiTEN-FF i uppgifter som speglar verkliga forskningsarbetsflöden. För läkemedelsliknande molekyler kan den optimera former så att de resulterande strukturerna nästan perfekt matchar dem från krävande kvantberäkningar, men tusentals gånger snabbare. I molekylär dynamik reproducerar den hur bindningslängder och vinklar fluktuerar över tid och följer kvantbaserade simuleringar tätt. Den utmärker sig också i att förutsäga hur energin förändras när en molekyl vrider sig runt en nyckelbindning, en avgörande komponent för att fånga konformationspreferenser i läkemedelsdesign. I flytande vatten och i korta peptider lösta i vatten genererar LiTEN-FF strukturella och termodynamiska egenskaper som stämmer väl överens med både experiment och dyrare referensmodeller, samtidigt som den levererar upp till tiofalt snabbare körning för stora system.
Föra sökandet efter användbara former framåt
Författarna demonstrerar också en praktisk konformer-sökningspipeline byggd kring LiTEN-FF. Genom upprepade cykler av höga temperaturer i dynamik, avkylning och snabb geometrioptimering genererar modellen rika mängder distinkta, lågenergetiska former för komplexa läkemedelsmolekyler. Jämfört med ett vida använt kvantbaserat arbetsflöde hittar detta AI-drivna angreppssätt mer diversifierade konformer på mindre än halva tiden. Dessutom, eftersom LiTEN-FF kan bearbeta många molekyler parallellt utan proportionell kostnadsökning, blir det särskilt kraftfullt för storskaliga screeningkampanjer där tusentals kandidater måste utvärderas.
Vad detta betyder för framtida läkemedels- och materialdesign
I grunden erbjuder LiTEN-FF en ny motor för molekylär simulering som för kvantnivåns tillförlitlighet mycket närmare daglig användbarhet. Genom att koda in geometrin hos vinklar och vridningar direkt i ett effektivt neuralt nätverk minskar den gapet mellan snabba men approximativa kraftfält och långsamma men precisa kvantberäkningar. För icke-specialister är slutsatsen att forskare snart kan komma att köra mycket mer realistiska molekylära ”experiment” på datorn, i skala och hastighet som är förenlig med modern läkemedelsforskning och materialutveckling. Om den antas i stor skala och förfinas ytterligare kan modeller i denna familj bli kärnkomponenter i automatiserade pipelines som föreslår, testar och förfinar nya molekyler långt innan de någon gång syntetiseras i labbet.
Citering: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Nyckelord: molekylär simulering, maskininlärningskraftfält, Läkemedelsupptäckt, biomolekylär modellering, kvantkemi