Clear Sky Science · it
Un modello di base scalabile e quantisticamente accurato per campi di forza biomolecolari tramite attenzione quadrangolare tensorizzata linearmente
Perché contano film molecolari più veloci
La scoperta di farmaci moderna e la progettazione dei materiali si basano sempre più su “film” al computer di molecole che si torcono, si flettono e reagiscono. Queste simulazioni possono rivelare come un farmaco si inserisce in una tasca proteica o come un nuovo materiale si comporta sotto stress. Ma gli strumenti odierni costringono gli scienziati a scegliere: metodi veloci che sacrificano accuratezza o calcoli quantistici ultra-precisi che sono troppo lenti per sistemi complessi e realistici. Questo articolo presenta LiTEN e il suo modello di campo di forza LiTEN-FF, un nuovo approccio di intelligenza artificiale che mira a offrire accuratezza a livello quantistico a velocità adatte alla modellizzazione molecolare quotidiana.
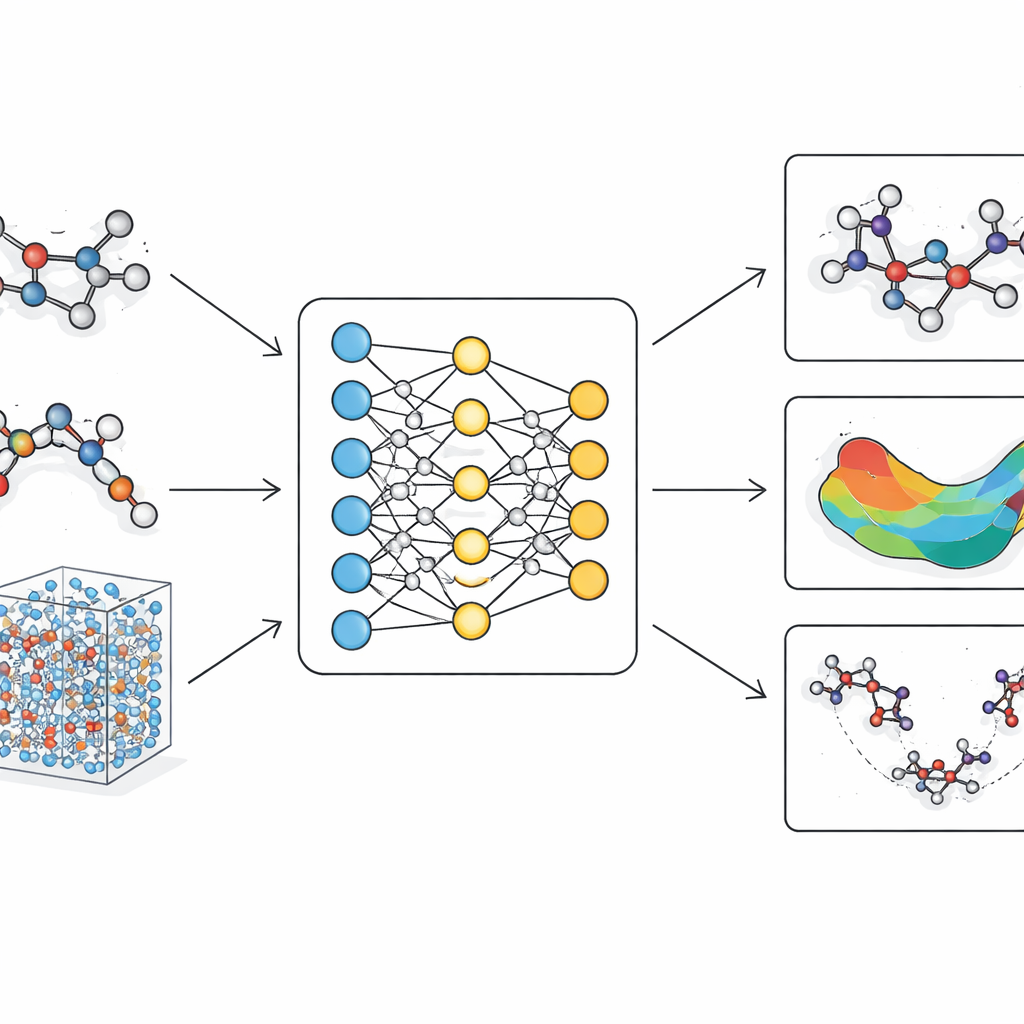
Il problema dei modelli molecolari attuali
Le simulazioni molecolari tradizionali si dividono in due categorie. I campi di forza classici trattano gli atomi come sfere connesse da molle con parametri fissi. Sono veloci e possono gestire grandi proteine o scale temporali estese, ma faticano a catturare sottili cambiamenti di forma, riarrangiamenti di legami e percorsi di reazione che contano per la chimica reale. I metodi quantistici, al contrario, descrivono esplicitamente gli elettroni e possono rappresentare con precisione la rottura e la formazione dei legami. Tuttavia sono così onerosi dal punto di vista computazionale che di solito sono limitati a molecole piccole o a simulazioni molto brevi. Negli ultimi anni sono apparsi approcci di apprendimento automatico come via di mezzo, che imparano a imitare i calcoli quantistici. Molti di questi modelli però mancano della rigore fisico necessario per essere affidabili o diventano troppo lenti quando scalati a biomolecole grandi e realistiche.
Un nuovo modo per insegnare all’IA le forme molecolari
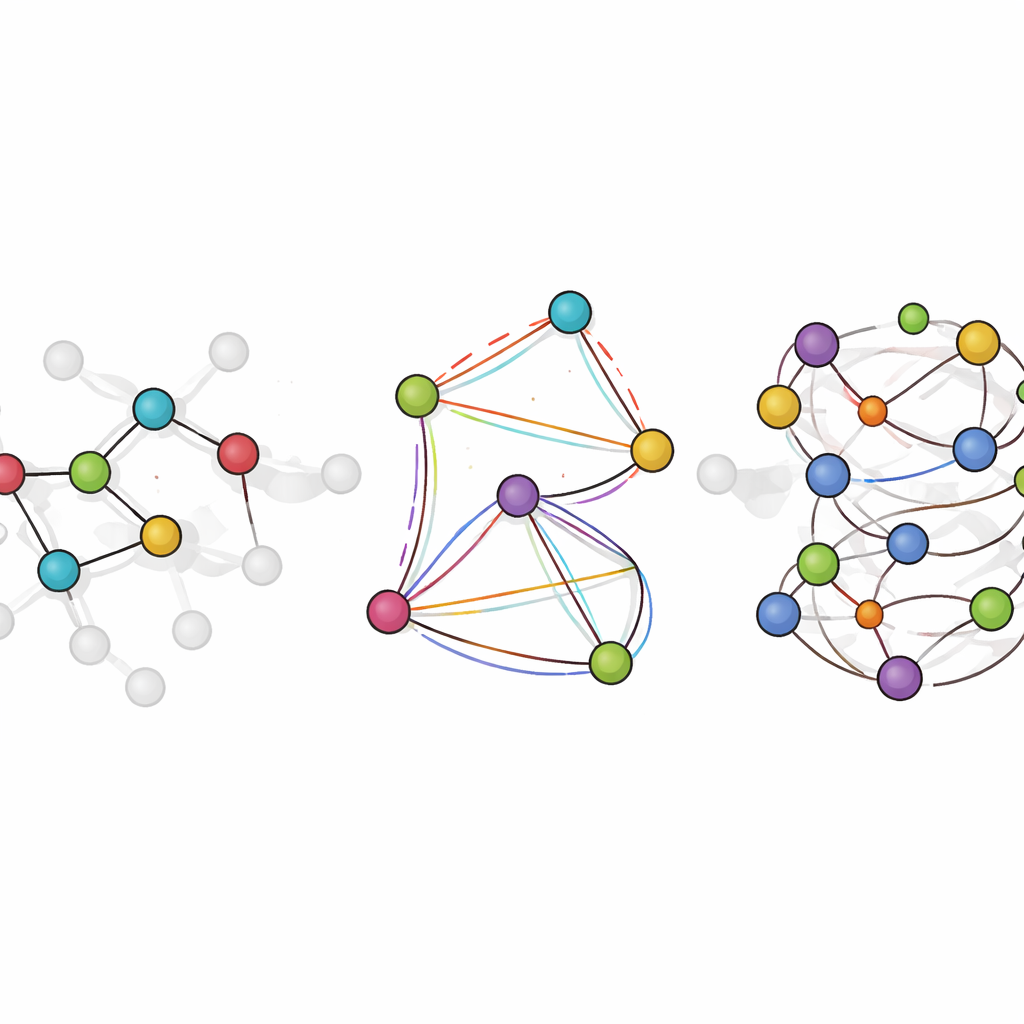
LiTEN affronta questa sfida riprogettando il modo in cui una rete neurale “percepisce” la geometria molecolare. Invece di considerare solo le semplici distanze a coppie tra atomi, LiTEN incorpora informazioni su pattern a tre e quattro atomi che governano angoli e torsioni nella molecola. È fondamentale che lo faccia rispettando le simmetrie fisiche di base: se ruoti o sposti una molecola nello spazio, l’energia prevista rimane la stessa e le forze previste ruotano in modo appropriato. L’innovazione chiave, chiamata attenzione quadrangolare tensorizzata, permette al modello di catturare interazioni complesse di piegamento e torsione usando operazioni vettoriali efficienti anziché matematica pesante e specialistica. Questo mantiene la computazione scalabile, così gli effetti molti-corpo che di solito appesantiscono i modelli avanzati possono essere gestiti con un costo che cresce solo linearmente con la dimensione del sistema.
Dall’addestramento su dati quantistici alle biomolecole reali
Sopra questa architettura, gli autori costruiscono LiTEN-FF, un “modello di base” per le forze molecolari. Lo addestrano innanzitutto su un enorme dataset di chimica quantistica contenente milioni di molecole con caratteristiche da farmaco, poi lo raffinano su una raccolta più piccola ma di maggiore accuratezza che include peptidi, strutture solvatate e elementi diversi come alogeni e metalli. Questo addestramento in due fasi consente al modello di apprendere sia una copertura chimica ampia sia i dettagli fini. Nei test di riferimento standard che confrontano energie e forze previste con risultati quantistici di alto livello, LiTEN eguaglia o supera i modelli di punta su molecole piccole e rigide e su sistemi biologicamente rilevanti molto più grandi. Mantiene l’accuratezza anche quando il numero di atomi cresce fino a centinaia, usando meno memoria e funzionando più rapidamente di molte alternative popolari.
Mettere il modello al lavoro nella chimica simulata
Oltre ai test statici, il team valuta LiTEN-FF in compiti che rispecchiano flussi di lavoro di ricerca reali. Per molecole simili a farmaci, può ottimizzare le conformazioni in modo che le strutture risultanti corrispondano quasi perfettamente a quelle ottenute con calcoli quantistici esigenti, ma migliaia di volte più velocemente. Nelle simulazioni di dinamica molecolare riproduce come lunghezze di legame e angoli fluttuano nel tempo, seguendo da vicino le simulazioni basate su metodi quantistici. Eccelle anche nel predire come l’energia cambia quando una molecola ruota attorno a un legame chiave, elemento cruciale per catturare le preferenze conformazionali nella progettazione di farmaci. In acqua liquida e in brevi peptidi disciolti in acqua, LiTEN-FF genera proprietà strutturali e termodinamiche in buon accordo sia con esperimenti sia con modelli di riferimento più costosi, offrendo allo stesso tempo fino a un’accelerazione di dieci volte sui sistemi grandi.
Velocizzare la ricerca di forme utili
Gli autori dimostrano anche una pipeline pratica di ricerca dei conformeri costruita attorno a LiTEN-FF. Eseguendo cicli ripetuti di dinamica ad alta temperatura, raffreddamento e rapido perfezionamento della geometria, il modello genera ricche serie di forme distinte a bassa energia per molecole complesse da farmaco. Rispetto a un flusso di lavoro quantum-based ampiamente usato, questo approccio guidato dall’IA trova conformeri più diversi in meno della metà del tempo. Inoltre, poiché LiTEN-FF può processare molte molecole in parallelo senza un aumento proporzionale del costo, diventa particolarmente potente per campagne di screening su larga scala dove migliaia di candidati devono essere valutati.
Cosa significa per il futuro della progettazione di farmaci e materiali
In sostanza, LiTEN-FF offre un nuovo motore per la simulazione molecolare che avvicina molto l’affidabilità a livello quantistico all’usabilità quotidiana. Incorpora la geometria di angoli e torsioni direttamente in una rete neurale efficiente, riducendo il divario tra campi di forza veloci ma approssimati e calcoli quantistici lenti ma accurati. Per i non specialisti, il messaggio è che i ricercatori potrebbero presto essere in grado di eseguire esperimenti molecolari al computer molto più realistici, a scale e velocità compatibili con la scoperta di farmaci e lo sviluppo di materiali moderni. Se adottati su larga scala e ulteriormente perfezionati, modelli di questa famiglia potrebbero diventare componenti chiave di pipeline automatizzate che propongono, testano e raffinano nuove molecole molto prima che vengano sintetizzate in laboratorio.
Citazione: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Parole chiave: simulazione molecolare, campi di forza basati su apprendimento automatico, scoperta di farmaci, modellizzazione biomolecolare, chimica quantistica