Clear Sky Science · nl
Een schaalbaar en kwantum-accurate fundatiemodel voor biomoleculaire krachtvelden via lineair getensoriseerde vierhoek-attentie
Waarom snellere moleculaire films ertoe doen
Moderne medicijnontdekking en materiaalontwerp zijn steeds vaker afhankelijk van computergestuurde “films” van moleculen die draaien, buigen en reageren. Deze simulaties kunnen laten zien hoe een geneesmiddel zich nestelt in een eiwitbinding of hoe een nieuw materiaal zich gedraagt onder belasting. Maar de huidige instrumenten dwingen wetenschappers tot een keuze: snelle methoden die ten koste van nauwkeurigheid vereenvoudigen, of ultra-precieze kwantumberekeningen die veel te traag zijn voor realistische, complexe systemen. Dit artikel introduceert LiTEN en het bijbehorende krachtveldmodel LiTEN-FF, een nieuwe kunstmatige-intelligentiebenadering die tot doel heeft kwantumniveau-nauwkeurigheid te leveren met snelheden die geschikt zijn voor alledaags moleculair modelleren.
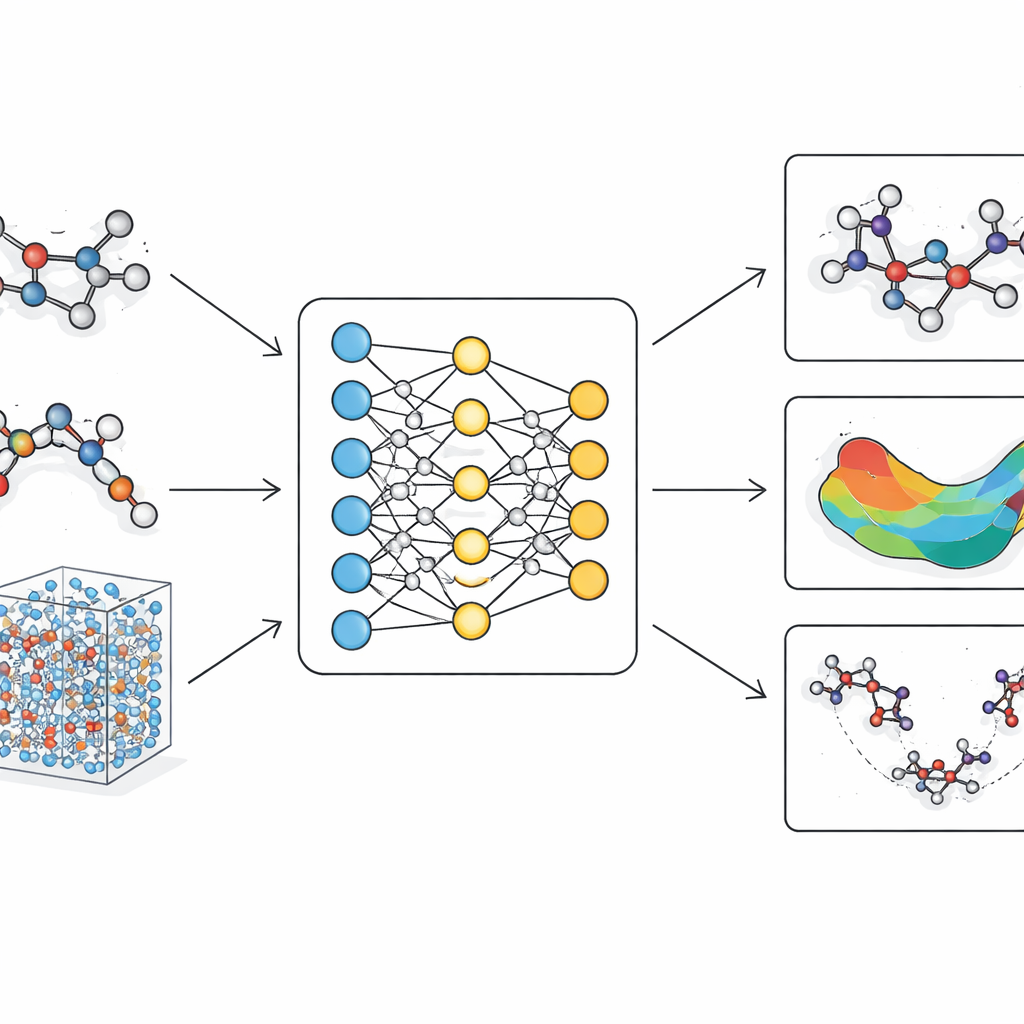
Het probleem met de huidige moleculaire modellen
Traditionele moleculaire simulaties vallen in twee kampen. Klassieke krachtvelden behandelen atomen als kralen die door veren zijn verbonden met vaste parameters. Ze draaien snel en kunnen grote eiwitten of lange tijdschalen aan, maar ze hebben moeite met subtiele vormveranderingen, bindingsherordening en reactiepaden die belangrijk zijn voor echte chemie. Kwantummethoden beschrijven daarentegen de elektronen expliciet en kunnen nauwkeurig het breken en vormen van bindingen vastleggen. Ze zijn echter zo berekeningsintensief dat ze meestal beperkt blijven tot kleine moleculen of zeer korte simulaties. In de afgelopen jaren zijn machine-learningbenaderingen verschenen als een middenweg, die leren kwantumberekeningen na te bootsen. Veel van deze modellen missen echter de fysieke degelijkheid die nodig is voor betrouwbaarheid, of ze worden te traag wanneer ze opgeschaald worden naar grote, realistische biomoleculen.
Een nieuwe manier om AI moleculaire vormen te laten begrijpen
LiTEN pakt deze uitdaging aan door te herontwerpen hoe een neuraal netwerk de moleculaire geometrie “voelt”. In plaats van alleen eenvoudige paarafstanden tussen atomen te beschouwen, bouwt LiTEN informatie in over patronen van drie en vier atomen die hoeken en torsies in een molecuul bepalen. Cruciaal is dat dit gebeurt met respect voor basale fysieke symmetrieën: als je een molecuul roteert of verplaatst, blijft de voorspelde energie gelijk en draaien de voorspelde krachten op de juiste manier mee. De belangrijkste innovatie, genoemd tensorized quadrangle attention (getensoriseerde vierhoek-attentie), stelt het model in staat complexe buig- en torsie-interacties vast te leggen met efficiënte vectorbewerkingen in plaats van zware, gespecialiseerde wiskunde. Dit houdt de berekening schaalbaar, zodat veeldeeltjeseffecten die geavanceerde modellen gewoonlijk vertragen met een kostenstijging die slechts lineair is in de systeemgrootte kunnen worden behandeld.
Van trainen op kwantumgegevens naar echte biomoleculen
Bovenop deze architectuur bouwen de auteurs LiTEN-FF, een “fundatiemodel” voor moleculaire krachten. Ze trainen het eerst op een enorme kwantumchemiedataset van miljoenen geneesmiddelachtige moleculen en verfijnen het vervolgens op een kleinere maar hogere-nauwkeurigheidsverzameling die peptiden, gesolvateerde structuren en diverse elementen zoals halogenen en metalen bevat. Deze tweefasen-training laat het model zowel brede chemische dekking als fijne details leren. In standaard benchmarktests die voorspelde energieën en krachten vergelijken met hoogwaardig kwantumreferenties, evenaart of overtreft LiTEN toonaangevende modellen voor zowel kleine, stijve moleculen als veel grotere, biologisch relevante systemen. Het behoudt nauwkeurigheid zelfs wanneer het aantal atomen in de honderden loopt, terwijl het minder geheugen gebruikt en sneller draait dan veel populaire alternatieven.
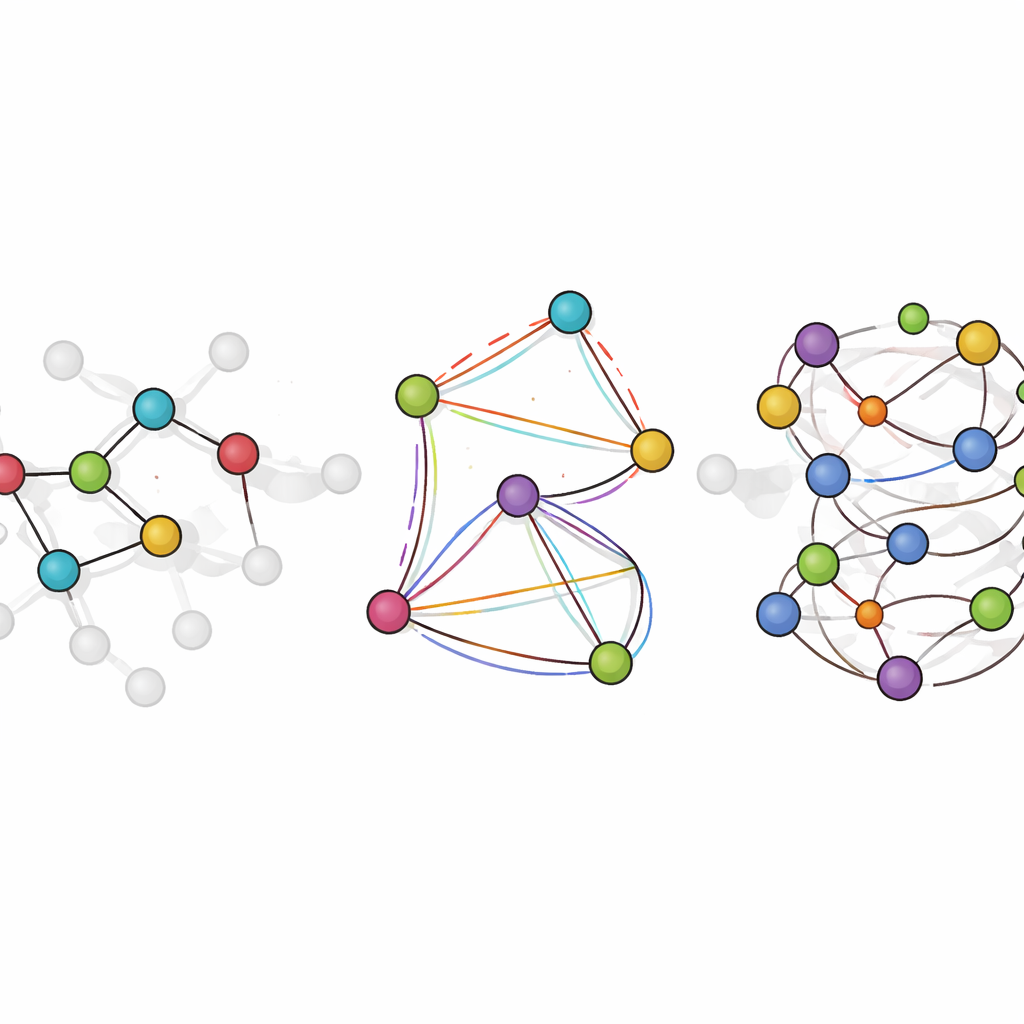
Het model inzetten in gesimuleerde chemie
Buiten statische tests evalueert het team LiTEN-FF in taken die echte onderzoeksworkflows nabootsen. Voor geneesmiddelachtige moleculen kan het vormen optimaliseren zodat de resulterende structuren bijna perfect overeenkomen met die van veeleisende kwantumberekeningen, maar duizenden keren sneller. In moleculaire dynamica runs reproduceert het hoe bindingslengten en hoeken in de tijd fluctueren en volgt het kwantumgebaseerde simulaties nauwgezet. Het blinkt ook uit in het voorspellen van energieveranderingen wanneer een molecuul rond een sleutelbinding draait, een cruciale factor voor het vastleggen van conformationele voorkeuren in medicijnontwerp. In vloeibaar water en in korte peptiden opgelost in water genereert LiTEN-FF structurele en thermodynamische eigenschappen die goed overeenkomen met zowel experimenten als duurdere referentiemodellen, terwijl het tot een tienvoudige versnelling biedt bij grote systemen.
Het versnellen van de zoektocht naar nuttige vormen
De auteurs demonstreren ook een praktische conformerzoekpipeline gebouwd rond LiTEN-FF. Door herhaalde cycli van hoge-temperatuur dynamica, afkoeling en snelle geometrieverfijning uit te voeren, genereert het model rijke verzamelingen van onderscheidende, energiearme vormen voor complexe geneesmiddelmoleculen. Vergeleken met een veelgebruikte kwantumgebaseerde workflow vindt deze AI-gestuurde benadering meer diverse conformeren in minder dan de helft van de tijd. Bovendien, doordat LiTEN-FF veel moleculen parallel kan verwerken zonder een evenredige stijging van de kosten, wordt het bijzonder krachtig voor grootschalige screeningscampagnes waarbij duizenden kandidaten geëvalueerd moeten worden.
Wat dit betekent voor toekomstig geneesmiddel- en materiaalontwerp
In wezen biedt LiTEN-FF een nieuwe motor voor moleculaire simulatie die kwantumniveau-betrouwbaarheid veel dichter bij dagelijkse bruikbaarheid brengt. Door de geometrie van hoeken en torsies direct in een efficiënt neuraal netwerk te coderen, verkleint het de kloof tussen snelle maar benaderende krachtvelden en trage maar nauwkeurige kwantumberekeningen. Voor niet-specialisten is de kernboodschap dat onderzoekers binnenkort veel realistischere moleculaire “experimenten” op de computer zouden kunnen uitvoeren, op schalen en met snelheden die compatibel zijn met moderne medicijnontdekking en materiaalontwikkeling. Als deze modellen breed worden aangenomen en verder verfijnd, zouden ze kerncomponenten kunnen worden van geautomatiseerde pipelines die nieuwe moleculen voorstellen, testen en verfijnen lang voordat ze ooit in het laboratorium worden gesynthetiseerd.
Bronvermelding: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Trefwoorden: moleculaire simulatie, machine learning-krachtvelden, medicijnontdekking, biomoleculair modelleren, kwantumchemie