Clear Sky Science · en
A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention
Why Faster Molecular Movies Matter
Modern drug discovery and materials design increasingly rely on computer “movies” of molecules twisting, flexing, and reacting. These simulations can reveal how a drug nestles into a protein pocket or how a new material behaves under stress. But today’s tools force scientists to choose: fast methods that cut corners on accuracy, or ultra-precise quantum calculations that are far too slow for realistic, complex systems. This paper introduces LiTEN and its force-field model LiTEN-FF, a new artificial intelligence approach that aims to deliver quantum-level accuracy at speeds suitable for everyday molecular modeling.
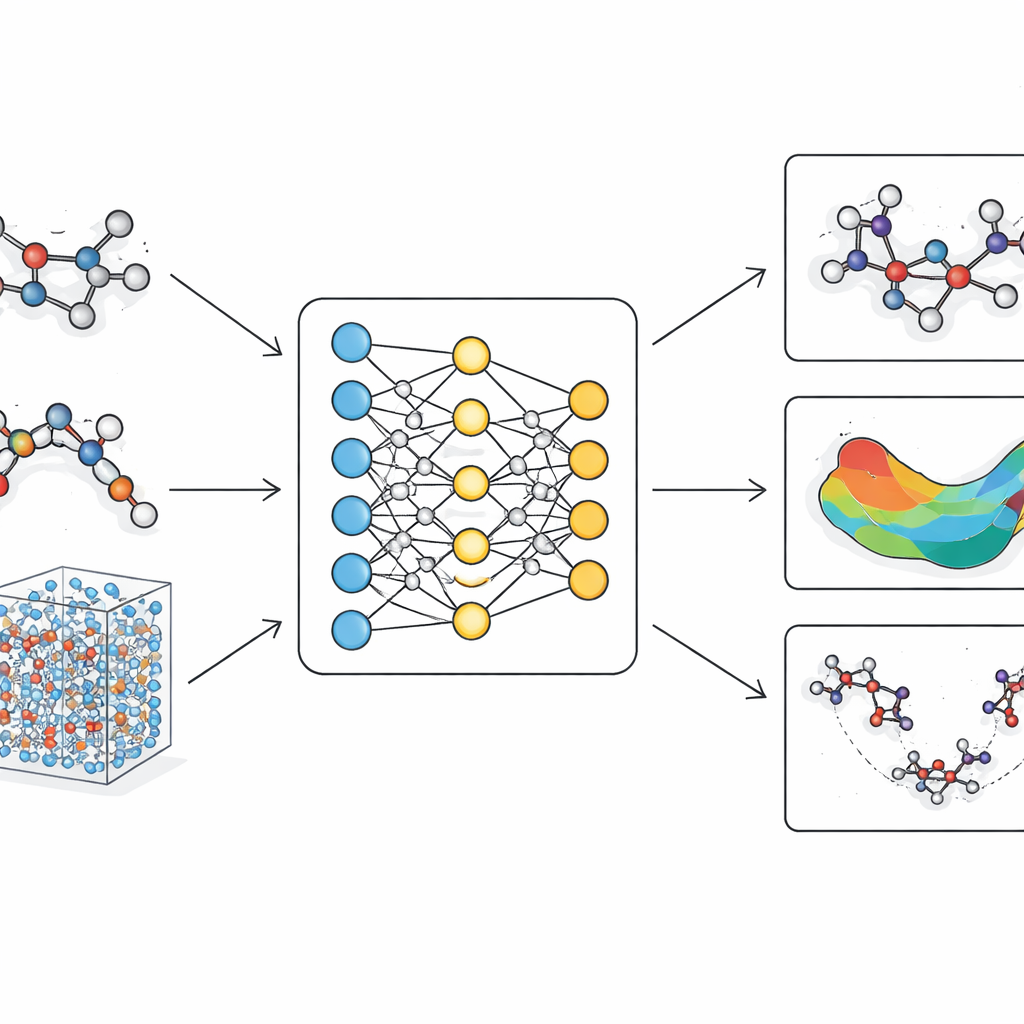
The Problem With Today’s Molecular Models
Traditional molecular simulations fall into two camps. Classical force fields treat atoms as beads connected by springs with fixed parameters. They run quickly and can handle large proteins or long time scales, but they struggle with subtle shape changes, bond rearrangements, and reaction pathways that matter for real chemistry. Quantum methods, in contrast, describe electrons explicitly and can accurately capture bond breaking and forming. However, they are so computationally demanding that they are usually limited to small molecules or very short simulations. Over the past few years, machine learning approaches have appeared as a middle way, learning to mimic quantum calculations. Yet many of these models either lack the physical rigor needed for reliability or become too slow when scaled to large, realistic biomolecules.
A New Way to Teach AI About Molecular Shapes
LiTEN tackles this challenge by redesigning how a neural network “feels” molecular geometry. Instead of only considering simple pairwise distances between atoms, LiTEN builds in information about three- and four-atom patterns that control angles and twists in a molecule. Crucially, it does this while respecting basic physical symmetries: if you rotate or move a molecule in space, its predicted energy stays the same, and the predicted forces rotate appropriately. The key innovation, called tensorized quadrangle attention, lets the model capture complex bending and twisting interactions using efficient vector operations rather than heavy, specialized mathematics. This keeps the computation scalable, so many-body effects that usually bog down advanced models can be handled with a cost that grows only linearly with system size.
From Training on Quantum Data to Real Biomolecules
On top of this architecture, the authors build LiTEN-FF, a “foundation model” for molecular forces. They first train it on a massive quantum chemistry dataset of millions of drug-like molecules, then refine it on a smaller but higher-accuracy collection that includes peptides, solvated structures, and diverse elements such as halogens and metals. This two-stage training lets the model learn both broad chemical coverage and fine detail. In standard benchmark tests that compare predicted energies and forces to high-level quantum results, LiTEN matches or surpasses leading models across small, rigid molecules and much larger, biologically relevant systems. It maintains accuracy even as the number of atoms grows into the hundreds, while using less memory and running faster than many popular alternatives.
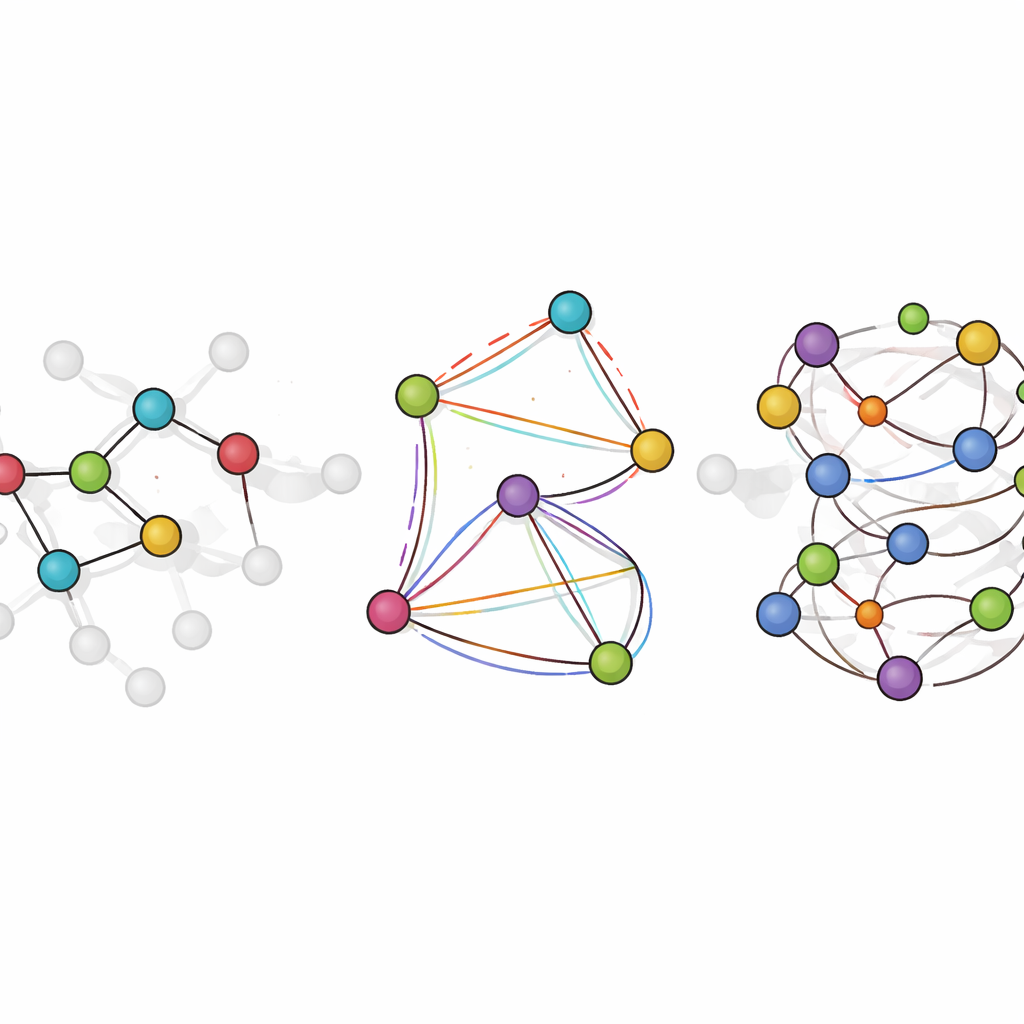
Putting the Model to Work in Simulated Chemistry
Beyond static tests, the team evaluates LiTEN-FF in tasks that mirror real research workflows. For drug-like molecules, it can optimize shapes so that the resulting structures almost perfectly match those from demanding quantum calculations, but thousands of times faster. In molecular dynamics runs, it reproduces how bond lengths and angles fluctuate over time, closely tracking quantum-based simulations. It also excels at predicting how energy changes when a molecule twists around a key bond, a crucial ingredient for capturing conformational preferences in drug design. In liquid water and in short peptides dissolved in water, LiTEN-FF generates structural and thermodynamic properties that agree well with both experiments and more expensive reference models, while delivering up to a tenfold speedup on large systems.
Speeding Up the Search for Useful Shapes
The authors also demonstrate a practical conformer search pipeline built around LiTEN-FF. By running repeated cycles of high-temperature dynamics, cooling, and quick geometry refinement, the model generates rich sets of distinct, low-energy shapes for complex drug molecules. Compared with a widely used quantum-based workflow, this AI-driven approach finds more diverse conformers in less than half the time. Moreover, because LiTEN-FF can process many molecules in parallel without a proportional rise in cost, it becomes particularly powerful for large-scale screening campaigns where thousands of candidates must be evaluated.
What This Means for Future Drug and Materials Design
In essence, LiTEN-FF offers a new engine for molecular simulation that brings quantum-level reliability much closer to everyday usability. By encoding the geometry of angles and twists directly into an efficient neural network, it narrows the gap between fast but approximate force fields and slow but accurate quantum calculations. For non-specialists, the takeaway is that researchers may soon be able to run far more realistic molecular “experiments” on the computer, at scales and speeds compatible with modern drug discovery and materials development. If adopted widely and further refined, models in this family could become core components of automated pipelines that propose, test, and refine new molecules long before they are ever synthesized in the lab.
Citation: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Keywords: molecular simulation, machine learning force fields, drug discovery, biomolecular modeling, quantum chemistry