Clear Sky Science · de
Ein skalierbares und quantengenaues Fundamentmodell für biomolekulare Kraftfelder durch linear tensorisierte Quadranten-Attention
Warum schnellere molekulare Filme wichtig sind
Moderne Wirkstoffforschung und Materialdesign stützen sich zunehmend auf Computer-„Filme“ von Molekülen, die sich drehen, biegen und reagieren. Diese Simulationen können zeigen, wie ein Wirkstoff in eine Protein-Tasche einsinkt oder wie ein neues Material sich unter Belastung verhält. Bisher zwingen die verfügbaren Werkzeuge Forscher zu einer Wahl: schnelle Methoden, die bei der Genauigkeit Abstriche machen, oder hochpräzise Quantenberechnungen, die für realistische, komplexe Systeme viel zu langsam sind. Dieser Artikel stellt LiTEN und sein Kraftfeldmodell LiTEN-FF vor, einen neuen KI-Ansatz, der Quanten-Niveau Genauigkeit mit Geschwindigkeiten verbinden will, die für den Alltag der molekularen Modellierung geeignet sind.
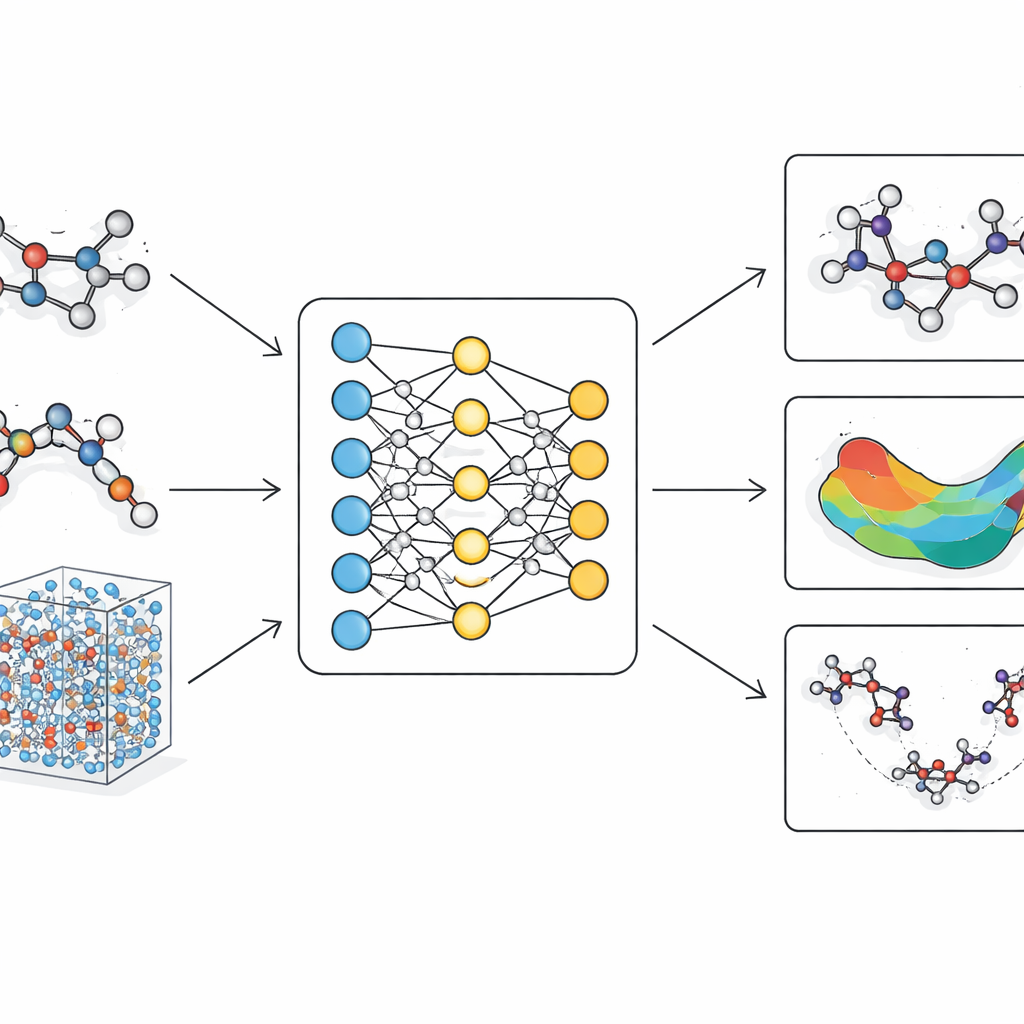
Das Problem heutiger molekularer Modelle
Traditionelle molekulare Simulationen fallen in zwei Kategorien. Klassische Kraftfelder behandeln Atome als Kugeln, verbunden durch Federn mit festen Parametern. Sie sind schnell und können große Proteine oder lange Zeitskalen verarbeiten, haben jedoch Schwierigkeiten mit subtilen Formänderungen, Bindungsumordnungen und Reaktionswegen, die für echte Chemie relevant sind. Quantenmethoden beschreiben dagegen Elektronen explizit und erfassen das Brechen und Bilden von Bindungen akkurat. Sie sind jedoch so rechenaufwendig, dass sie meist auf kleine Moleküle oder sehr kurze Simulationen beschränkt bleiben. In den letzten Jahren sind maschinelle Lernansätze als Mittelweg aufgetaucht, die lernen, Quantenberechnungen zu imitieren. Viele dieser Modelle fehlen jedoch die physikalische Strenge für zuverlässige Vorhersagen, oder sie werden zu langsam, wenn sie auf große, realistische Biomoleküle skaliert werden.
Eine neue Art, KI Molekülgeometrie beizubringen
LiTEN begegnet dieser Herausforderung, indem es neu gestaltet, wie ein neuronales Netzwerk molekulare Geometrie „fühlt“. Statt nur einfache paarweise Abstände zwischen Atomen zu betrachten, integriert LiTEN Informationen über Drei- und Vier-Atom-Muster, die Winkel und Verdrehungen in einem Molekül steuern. Entscheidend ist, dass es dabei grundlegende physikalische Symmetrien respektiert: Dreht oder verschiebt man ein Molekül im Raum, bleibt seine vorhergesagte Energie gleich, und die vorhergesagten Kräfte drehen sich entsprechend mit. Die Schlüsselinnovation, genannt tensorisierte Quadranten-Attention, erlaubt es dem Modell, komplexe Biege- und Verdrehungswechselwirkungen mit effizienten Vektoroperationen statt schwerer, spezialisierter Mathematik zu erfassen. Dadurch bleibt die Berechnung skalierbar, sodass Vielteilcheneffekte, die fortgeschrittene Modelle gewöhnlich ausbremsen, mit Kosten behandelt werden können, die nur linear mit der Systemgröße wachsen.
Vom Training an quantenchemischen Daten zu realen Biomolekülen
Auf dieser Architektur bauen die Autoren LiTEN-FF, ein „Fundamentmodell“ für molekulare Kräfte, auf. Zuerst trainieren sie es an einem massiven Datensatz aus der Quantenchemie mit Millionen wirkstoffähnlichen Molekülen, anschließend verfeinern sie es an einer kleineren, aber höher genauen Sammlung, die Peptide, solvatisierte Strukturen und verschiedene Elemente wie Halogene und Metalle enthält. Dieses zweistufige Training ermöglicht dem Modell, sowohl breite chemische Abdeckung als auch feine Details zu lernen. In Standard-Benchmark-Tests, die vorhergesagte Energien und Kräfte mit hochrangigen Quantenreferenzen vergleichen, erreicht LiTEN gleichwertige oder bessere Ergebnisse als führende Modelle – sowohl bei kleinen, starren Molekülen als auch bei deutlich größeren, biologisch relevanten Systemen. Es bewahrt die Genauigkeit auch, wenn die Anzahl der Atome in die Hunderte wächst, während es weniger Arbeitsspeicher benötigt und schneller läuft als viele verbreitete Alternativen.
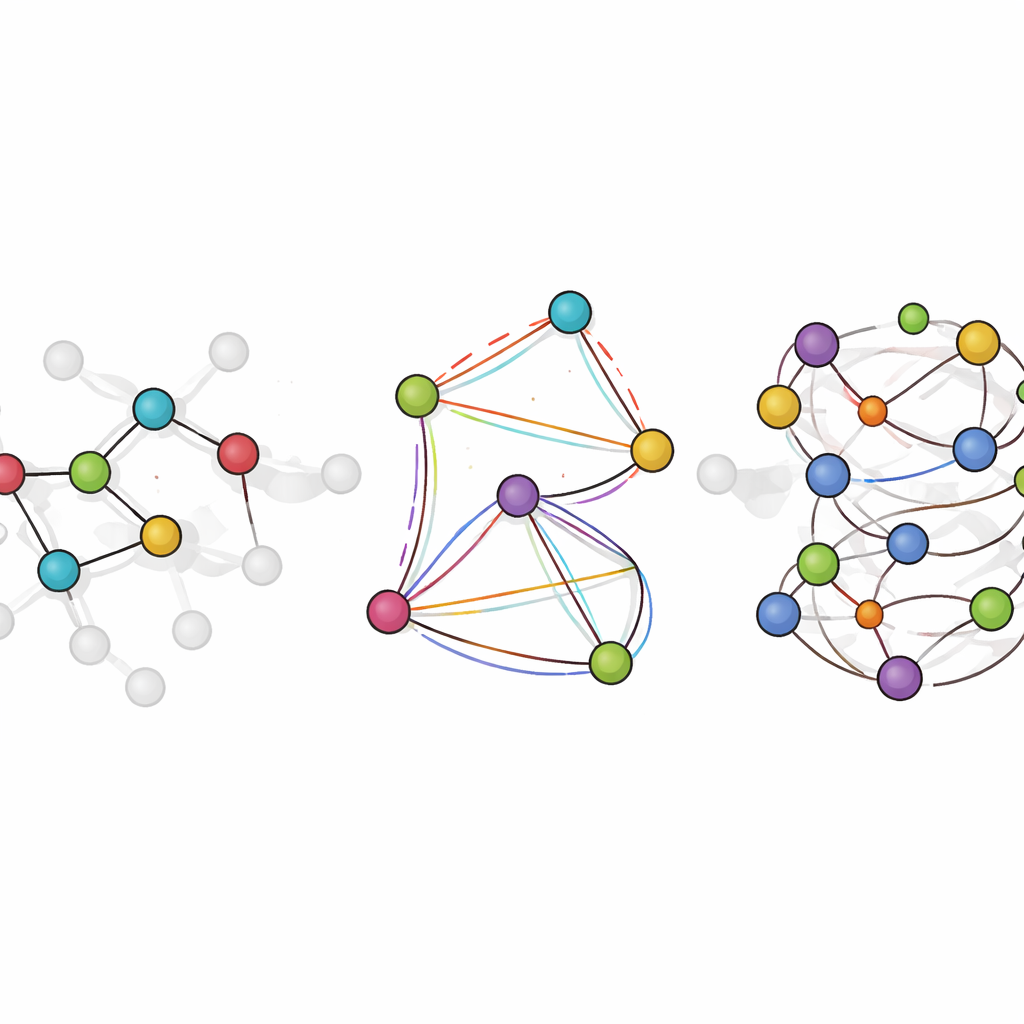
Das Modell in simulierter Chemie anwenden
Über statische Tests hinaus bewertet das Team LiTEN-FF in Aufgaben, die reale Forschungsabläufe nachbilden. Bei wirkstoffähnlichen Molekülen kann es Konformationen optimieren, sodass die resultierenden Strukturen nahezu perfekt mit aufwändigen Quantenberechnungen übereinstimmen – aber tausendfach schneller. In Molekulardynamik-Läufen reproduziert es, wie Bindungslängen und -winkel über die Zeit schwanken, und folgt Quanten-basierten Simulationen eng. Es ist auch besonders gut darin, zu prognostizieren, wie sich die Energie ändert, wenn ein Molekül um eine Schlüsselbindung verdreht wird – ein wichtiges Element, um Konformationspräferenzen im Wirkstoffdesign zu erfassen. In flüssigem Wasser und in kurzen, in Wasser gelösten Peptiden erzeugt LiTEN-FF strukturelle und thermodynamische Eigenschaften, die sowohl mit Experimenten als auch mit teureren Referenzmodellen gut übereinstimmen, und liefert gleichzeitig bis zu eine Verzehnfachung der Geschwindigkeit bei großen Systemen.
Die Suche nach nützlichen Formen beschleunigen
Die Autoren zeigen außerdem eine praktische Konformersuch-Pipeline, die auf LiTEN-FF basiert. Durch wiederholte Zyklen aus Hochtemperatur-Dynamik, Abkühlung und schneller Geometrieverfeinerung erzeugt das Modell reichhaltige Mengen unterschiedlicher, energiearmer Formen für komplexe Wirkstoffmoleküle. Im Vergleich zu einem weit verbreiteten quantenbasierten Workflow findet dieser KI-getriebene Ansatz vielfältigere Konformere in weniger als der Hälfte der Zeit. Da LiTEN-FF viele Moleküle parallel verarbeiten kann, ohne dass die Kosten proportional ansteigen, ist es besonders leistungsfähig für groß angelegte Screening-Kampagnen, bei denen Tausende von Kandidaten bewertet werden müssen.
Was das für die zukünftige Wirkstoff- und Materialentwicklung bedeutet
Im Kern bietet LiTEN-FF eine neue Engine für molekulare Simulationen, die die Quanten-Genauigkeit deutlich näher an den Alltag bringt. Indem die Geometrie von Winkeln und Verdrehungen direkt in ein effizientes neuronales Netzwerk kodiert wird, schließt es die Lücke zwischen schnellen, aber approximativen Kraftfeldern und langsamen, aber genauen Quantenberechnungen. Für Nicht-Spezialisten lautet die Kernbotschaft, dass Forscher möglicherweise bald weitaus realistischere molekulare „Experimente“ am Computer durchführen können, in Größenordnungen und Geschwindigkeiten, die mit moderner Wirkstoffforschung und Materialentwicklung kompatibel sind. Werden solche Modelle breit angenommen und weiter verfeinert, könnten sie zu zentralen Komponenten automatisierter Pipelines werden, die neue Moleküle vorschlagen, testen und verfeinern, lange bevor sie im Labor synthetisiert werden.
Zitation: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Schlüsselwörter: molekulare Simulation, maschinelles Lernen Kraftfelder, Wirkstoffforschung, biomolekulares Modellieren, Quantenchemie