Clear Sky Science · es
Un modelo básico escalable y con precisión cuántica para campos de fuerza biomoleculares mediante atención en cuadrángulos tensorizada linealmente
Por qué importan las películas moleculares más rápidas
El descubrimiento de fármacos y el diseño de materiales modernos dependen cada vez más de “películas” por ordenador que muestran moléculas torciéndose, flexionándose y reaccionando. Estas simulaciones pueden revelar cómo un fármaco se acomoda en la cavidad de una proteína o cómo se comporta un nuevo material bajo tensión. Pero las herramientas actuales obligan a los científicos a elegir: métodos rápidos que sacrifican precisión, o cálculos cuánticos ultra-precisos que son demasiado lentos para sistemas complejos y realistas. Este artículo presenta LiTEN y su modelo de campo de fuerza LiTEN-FF, un nuevo enfoque de inteligencia artificial que pretende ofrecer precisión a nivel cuántico con velocidades adecuadas para el modelado molecular cotidiano.
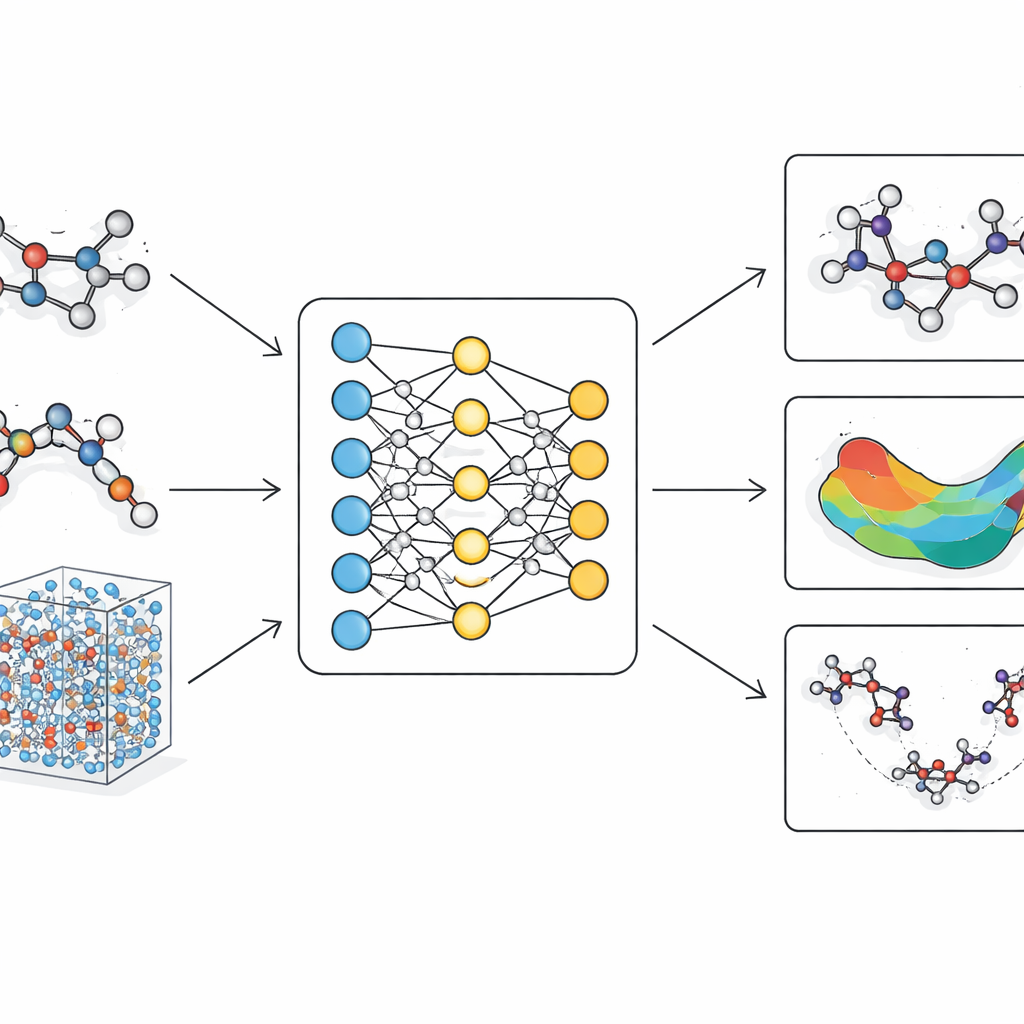
El problema de los modelos moleculares actuales
Las simulaciones moleculares tradicionales se dividen en dos bandos. Los campos de fuerza clásicos tratan los átomos como esferas conectadas por resortes con parámetros fijos. Son rápidos y pueden manejar grandes proteínas o escalas temporales largas, pero tienen dificultades con cambios sutiles de forma, reorganizaciones de enlaces y vías de reacción que importan en química real. Los métodos cuánticos, en contraste, describen los electrones explícitamente y pueden capturar con precisión la ruptura y formación de enlaces. Sin embargo, son tan costosos computacionalmente que normalmente se limitan a moléculas pequeñas o simulaciones muy cortas. En los últimos años han surgido enfoques de aprendizaje automático como camino intermedio, aprendiendo a imitar cálculos cuánticos. Aun así, muchos de estos modelos carecen del rigor físico necesario para ser fiables o se vuelven demasiado lentos cuando se escalan a biomoléculas grandes y realistas.
Una nueva forma de enseñar a la IA sobre las formas moleculares
LiTEN aborda este desafío rediseñando cómo una red neuronal “percibe” la geometría molecular. En lugar de considerar solo distancias par a par entre átomos, LiTEN incorpora información sobre patrones de tres y cuatro átomos que controlan ángulos y torsiones en una molécula. De forma crucial, lo hace respetando las simetrías físicas básicas: si rotas o mueves una molécula en el espacio, su energía predicha permanece igual y las fuerzas predichas rotan de forma adecuada. La innovación clave, llamada atención en cuadrángulos tensorizada, permite al modelo capturar interacciones complejas de flexión y torsión usando operaciones vectoriales eficientes en lugar de matemáticas pesadas y especializadas. Esto mantiene el cálculo escalable, de modo que los efectos muchos-cuerpos que suelen ralentizar modelos avanzados pueden abordarse con un coste que crece solo linealmente con el tamaño del sistema.
Del entrenamiento con datos cuánticos a biomoléculas reales
Sobre esta arquitectura, los autores construyen LiTEN-FF, un “modelo base” para fuerzas moleculares. Primero lo entrenan con un enorme conjunto de datos de química cuántica que contiene millones de moléculas tipo fármaco, y luego lo afinan con una colección más pequeña pero de mayor precisión que incluye péptidos, estructuras solvadas y elementos diversos como halógenos y metales. Este entrenamiento en dos fases permite al modelo aprender tanto una cobertura química amplia como los detalles finos. En pruebas estándar de referencia que comparan energías y fuerzas predichas con resultados cuánticos de alto nivel, LiTEN iguala o supera a los modelos líderes tanto en moléculas pequeñas y rígidas como en sistemas biológicamente relevantes mucho mayores. Mantiene la precisión incluso cuando el número de átomos crece hasta cientos, utilizando menos memoria y ejecutándose más rápido que muchas alternativas populares.
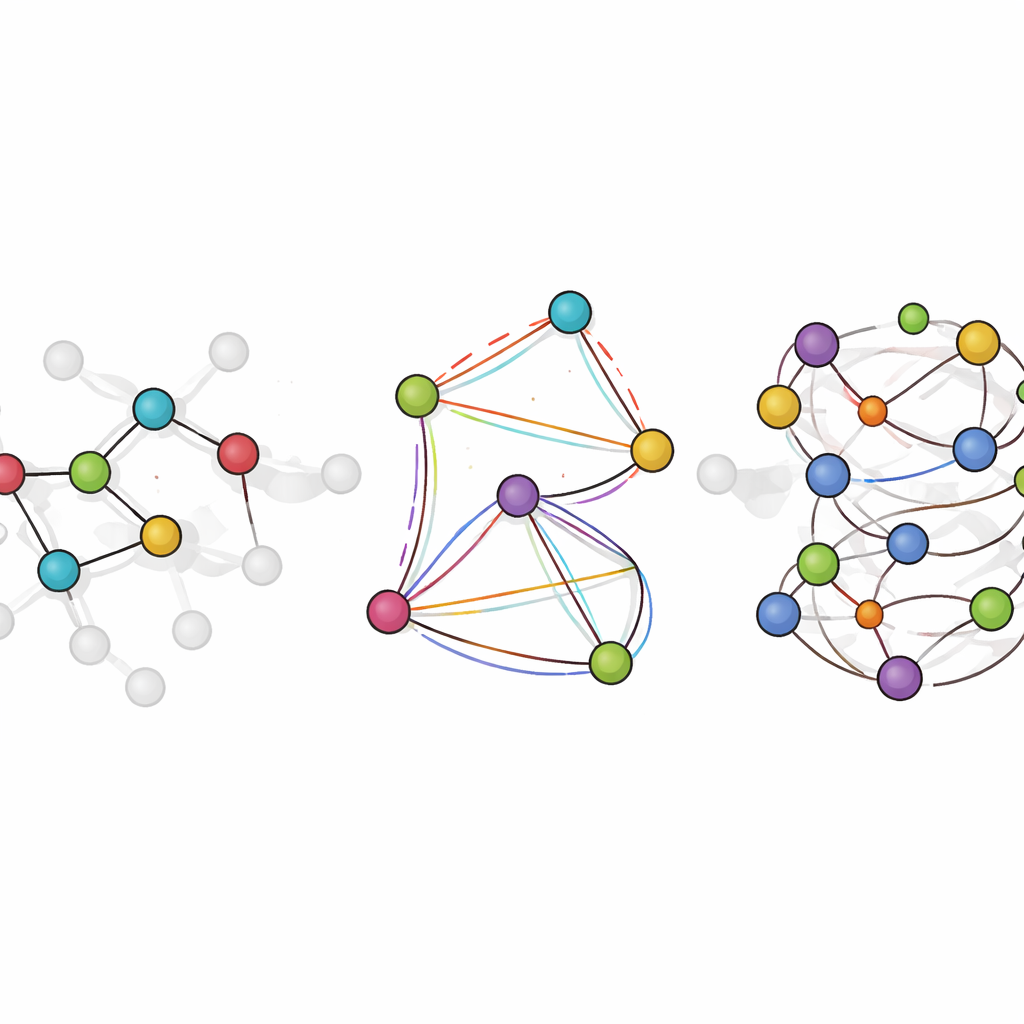
Poniendo el modelo a funcionar en química simulada
Más allá de pruebas estáticas, el equipo evalúa LiTEN-FF en tareas que reflejan flujos de trabajo de investigación reales. Para moléculas tipo fármaco, puede optimizar formas de modo que las estructuras resultantes coincidan casi perfectamente con las de cálculos cuánticos exigentes, pero miles de veces más rápido. En simulaciones de dinámica molecular, reproduce cómo fluctúan longitudes de enlace y ángulos con el tiempo, siguiendo de cerca las simulaciones basadas en mecánica cuántica. También sobresale al predecir cómo cambia la energía cuando una molécula gira alrededor de un enlace clave, un ingrediente crítico para captar preferencias conformacionales en diseño de fármacos. En agua líquida y en péptidos cortos disueltos en agua, LiTEN-FF genera propiedades estructurales y termodinámicas que concuerdan bien con experimentos y con modelos de referencia más costosos, al tiempo que ofrece hasta un aceleramiento de diez veces en sistemas grandes.
Acelerar la búsqueda de formas útiles
Los autores también demuestran una canalización práctica de búsqueda de conformadores basada en LiTEN-FF. Ejecutando ciclos repetidos de dinámica a alta temperatura, enfriamiento y refinamiento geométrico rápido, el modelo genera conjuntos ricos de formas distintas y de baja energía para moléculas farmacológicas complejas. En comparación con un flujo de trabajo cuántico ampliamente utilizado, este enfoque impulsado por IA encuentra conformadores más diversos en menos de la mitad del tiempo. Además, dado que LiTEN-FF puede procesar muchas moléculas en paralelo sin un aumento proporcional del coste, resulta especialmente potente para campañas de cribado a gran escala en las que deben evaluarse miles de candidatos.
Qué significa esto para el futuro del diseño de fármacos y materiales
En esencia, LiTEN-FF ofrece un nuevo motor para la simulación molecular que acerca la fiabilidad a nivel cuántico a la usabilidad cotidiana. Al codificar la geometría de ángulos y torsiones directamente en una red neuronal eficiente, reduce la brecha entre campos de fuerza rápidos pero aproximados y cálculos cuánticos lentos pero precisos. Para quienes no son especialistas, la conclusión es que los investigadores pronto podrán ejecutar muchos más “experimentos” moleculares realistas por ordenador, a escalas y velocidades compatibles con el descubrimiento moderno de fármacos y el desarrollo de materiales. Si se adopta ampliamente y se refina aún más, los modelos de esta familia podrían convertirse en componentes centrales de tuberías automatizadas que proponen, prueban y refinan nuevas moléculas mucho antes de que se sinteticen en el laboratorio.
Cita: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Palabras clave: simulación molecular, campos de fuerza por aprendizaje automático, descubrimiento de fármacos, modelado biomolecular, química cuántica