Clear Sky Science · ru
Масштабируемая и квантово-точная «фундаментальная» модель для биомолекулярных силовых полей через линейно тензоризованное четырехугольное внимание
Почему важны более быстрые молекулярные «фильмы»
Современный поиск лекарств и проектирование материалов всё чаще опираются на компьютерные «кинофильмы» молекул, показывающие, как они крутятся, изгибаются и реагируют. Такие симуляции позволяют понять, как лекарство устраивается в кармане белка или как новый материал ведёт себя при нагрузке. Но нынешние инструменты вынуждают учёных выбирать: либо быстрые методы с компромиссами по точности, либо ультраточные квантовые расчёты, которые слишком медленны для реалистичных сложных систем. В этой работе представлена LiTEN и её силевая модель LiTEN-FF — новый подход на основе искусственного интеллекта, который стремится обеспечить квантовый уровень точности при скоростях, пригодных для повседневного молекулярного моделирования.
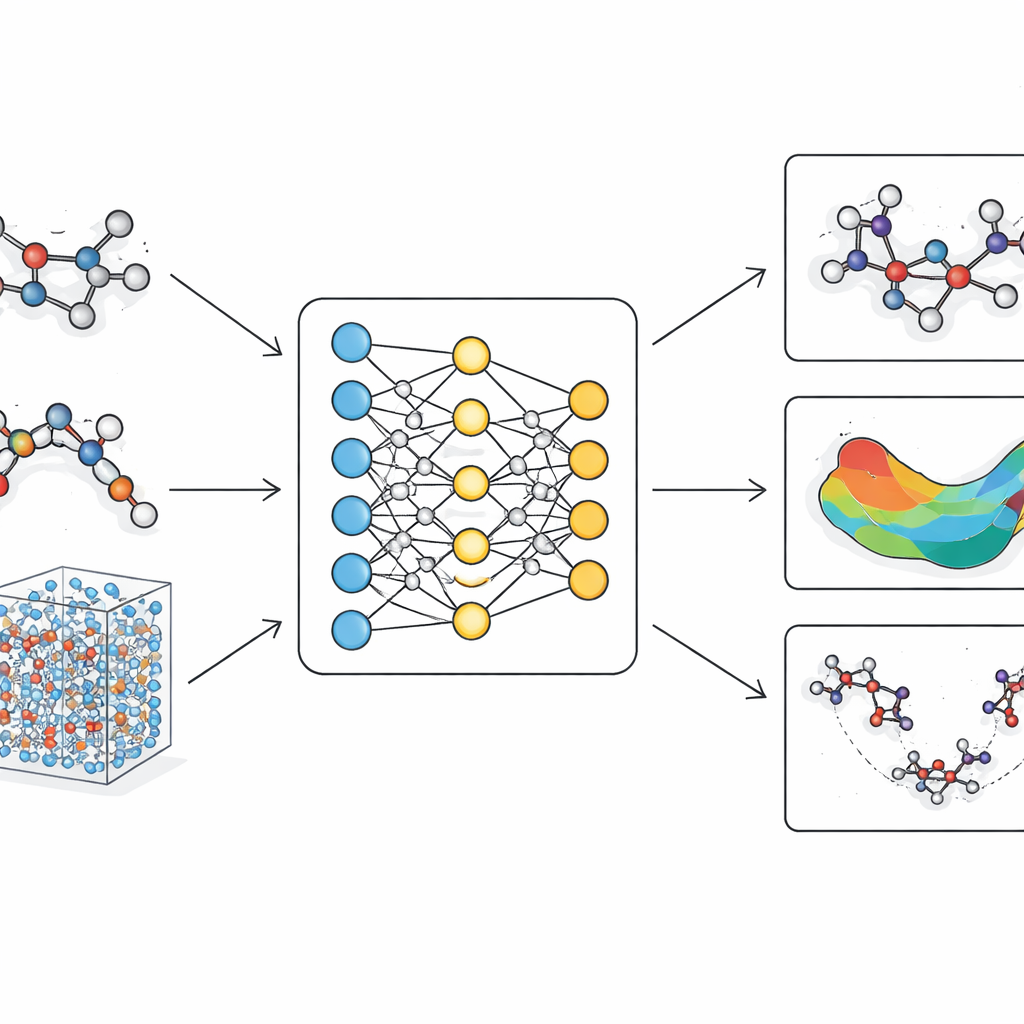
В чём проблема современных молекулярных моделей
Традиционные молекулярные симуляции делятся на два лагеря. Классические силовые поля рассматривают атомы как шарики, соединённые пружинами с фиксированными параметрами. Они работают быстро и способны обрабатывать большие белки или длинные временные масштабы, но с трудом описывают тонкие изменения формы, перестановки связей и реакционные пути, важные для реальной химии. Квантовые методы, напротив, явно описывают электроны и могут точно фиксировать разрыв и образование связей. Однако они настолько ресурсоёмки, что обычно ограничиваются малыми молекулами или очень короткими симуляциями. За последние годы появились подходы на основе машинного обучения как нечто среднее, подражающее квантовым расчётам. Тем не менее многие из этих моделей либо лишены необходимой физической строгости для надёжности, либо становятся слишком медленными при масштабировании до крупных, реалистичных биомолекул.
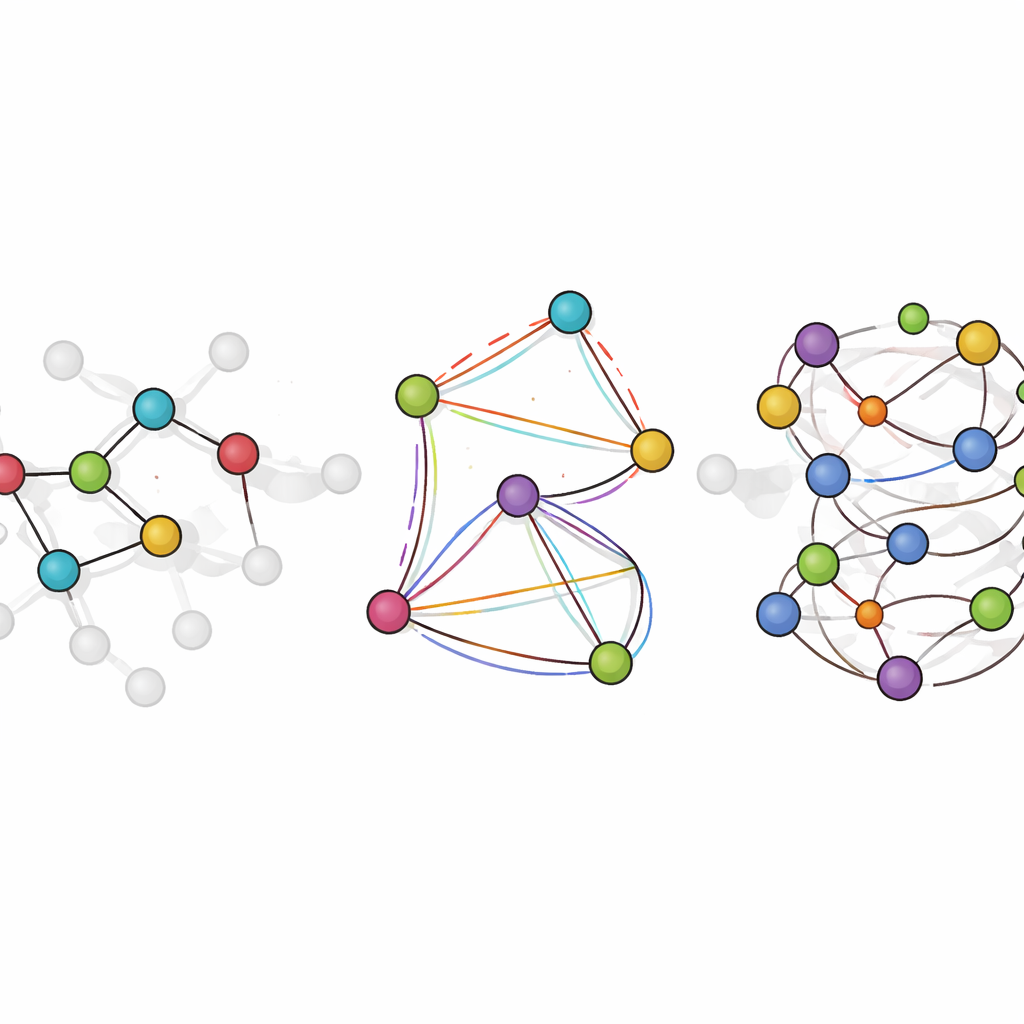
Новый способ научить ИИ понимать молекулярные формы
LiTEN решает эту задачу, перестраивая то, как нейронная сеть «ощущает» молекулярную геометрию. Вместо того чтобы учитывать только простые попарные расстояния между атомами, LiTEN включает информацию о шаблонах из трёх и четырёх атомов, которые контролируют углы и скрутки в молекуле. Важно, что это делается с соблюдением базовых физических симметрий: при вращении или переносе молекулы в пространстве предсказанная энергия остаётся неизменной, а предсказанные силы правильно трансформируются. Ключевая новация, называемая тензоризованным четырехугольным вниманием, позволяет модели захватывать сложные изгибы и кручения с помощью эффективных векторных операций вместо тяжёлой специализированной математики. Это сохраняет вычисления масштабируемыми, так что эффекты многих тел, которые обычно замедляют продвинутые модели, обрабатываются с затратой, растущей лишь линейно с размером системы.
От обучения на квантовых данных к реальным биомолекулам
На базе этой архитектуры авторы построили LiTEN-FF — «фундаментальную модель» сил. Сначала её обучают на массивном наборе данных квантовой химии, содержащем миллионы молекул, похожих на лекарственные вещества, а затем дообучают на более компактной, но более точной коллекции, включающей пептиды, растворённые структуры и разнообразные элементы, такие как галогены и металлы. Эта двухэтапная подготовка позволяет модели освоить как широкий химический охват, так и тонкие детали. В стандартных тестах-бенчмарках, сравнивающих предсказанные энергии и силы с высокоуровневыми квантовыми результатами, LiTEN соответствует или превосходит ведущие модели как на малых жёстких молекулах, так и на значительно больших биологически релевантных системах. Она сохраняет точность даже при сотнях атомов, потребляет меньше памяти и работает быстрее многих популярных альтернатив.
Применение модели в моделируемой химии
Помимо статических тестов, команда оценивает LiTEN-FF в задачах, приближённых к реальным исследовательским рабочим процессам. Для молекул, похожих на лекарственные, она может оптимизировать конформации так, что полученные структуры почти идеально совпадают с результатами требовательных квантовых расчётов, но в тысячи раз быстрее. В задачах молекулярной динамики модель воспроизводит флуктуации длин связей и углов во времени, близко следуя квантовым симуляциям. Она также превосходно предсказывает изменение энергии при повороте вокруг ключевой связи — важный фактор для захвата конформационных предпочтений в дизайне лекарств. В жидкой воде и в коротких пептидах, растворённых в воде, LiTEN-FF генерирует структурные и термодинамические свойства, хорошо согласующиеся как с экспериментами, так и с более дорогими эталонными моделями, обеспечивая при этом до десятикратного ускорения для больших систем.
Ускорение поиска полезных форм
Авторы также демонстрируют практический конформерный поисковый конвейер на базе LiTEN-FF. Запуская повторяющиеся циклы высокотемпературной динамики, охлаждения и быстрой доработки геометрии, модель генерирует богатые наборы различных низкоэнергетических форм для сложных лекарственных молекул. По сравнению с широко используемым квантовым рабочим процессом, этот подход на базе ИИ находит более разнообразные конформеры менее чем за половину времени. Более того, поскольку LiTEN-FF может обрабатывать многие молекулы параллельно без пропорционального роста затрат, она особенно эффективна для масштабных скрининговых кампаний, где нужно оценить тысячи кандидатов.
Что это значит для будущего разработки лекарств и материалов
По сути, LiTEN-FF предлагает новый «движок» для молекулярного моделирования, который делает квантовый уровень надёжности гораздо ближе к повседневной применимости. Кодируя геометрию углов и скруток непосредственно в эффективную нейронную сеть, модель сокращает разрыв между быстрыми, но приближенными силовыми полями и медленными, но точными квантовыми расчётами. Для неспециалистов главное — исследователи вскоре смогут запускать значительно более реалистичные молекулярные «эксперименты» на компьютере, в масштабах и с скоростью, совместимыми с современным поиском лекарств и разработкой материалов. При широком принятии и дальнейшем совершенствовании модели этого семейства могут стать основными компонентами автоматизированных конвейеров, которые предлагают, тестируют и уточняют новые молекулы задолго до их синтеза в лаборатории.
Цитирование: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Ключевые слова: молекулярное моделирование, машинно-обучаемые силовые поля, поиск лекарств, биомолекулярное моделирование, квантовая химия