Clear Sky Science · pl
Skalowalny i zgodny z mechaniką kwantową model podstawowy dla pól sił biomolekularnych poprzez liniowo tensoryzowaną uwagę czworokątną
Dlaczego szybsze „filmy” molekularne mają znaczenie
Współczesne odkrywanie leków i projektowanie materiałów coraz częściej opierają się na komputerowych „filmach” pokazujących, jak molekuły skręcają się, wyginają i reagują. Takie symulacje mogą ujawnić, jak lek osiada w kieszeni białka lub jak nowy materiał zachowuje się pod obciążeniem. Jednak dostępne narzędzia zmuszają naukowców do wyboru: szybkie metody, które upraszczają złożoność, albo ultradokładne obliczenia kwantowe, zbyt wolne dla realistycznych, złożonych układów. W artykule przedstawiono LiTEN i jego model pola sił LiTEN-FF — nowe podejście oparte na sztucznej inteligencji, które ma dostarczać dokładności na poziomie kwantowym przy prędkościach odpowiednich do codziennego modelowania molekularnego.
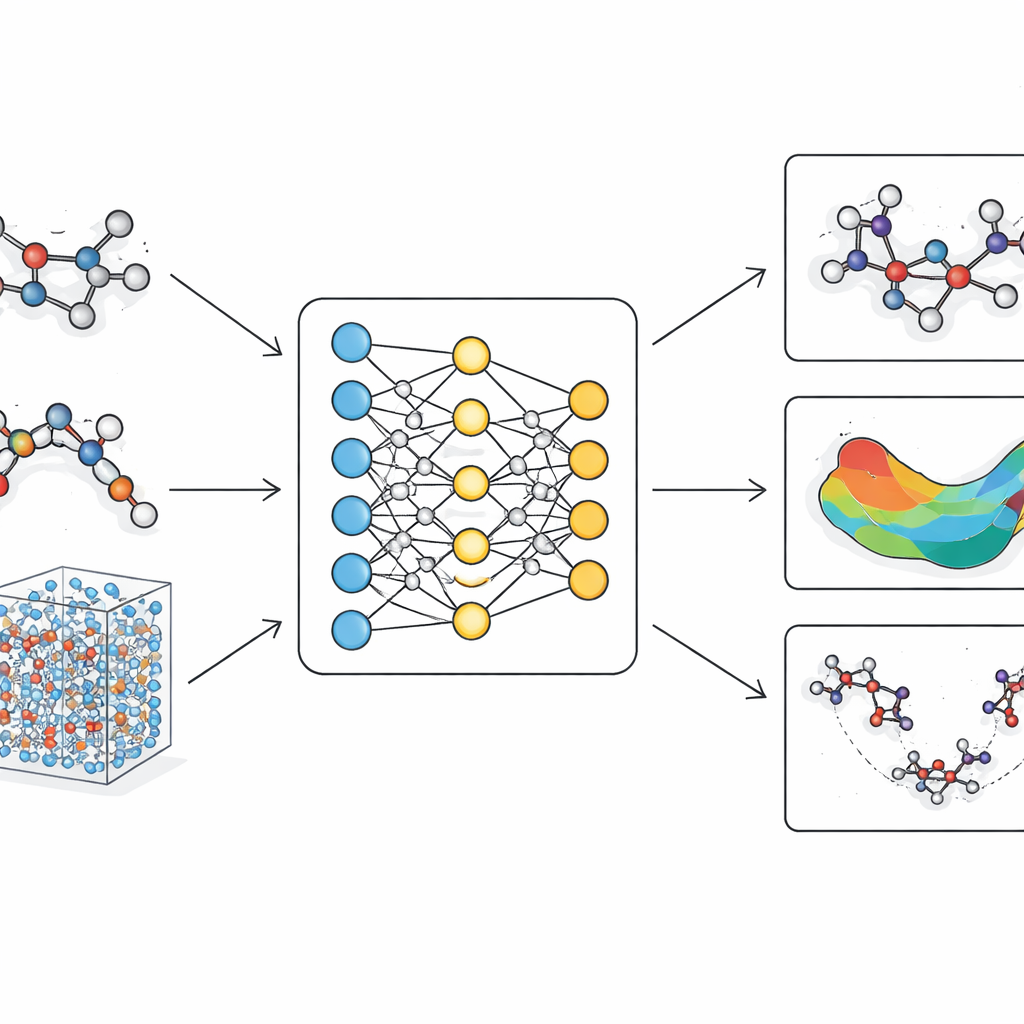
Problem dzisiejszych modeli molekularnych
Tradycyjne symulacje molekularne dzielą się na dwie grupy. Klasyczne pola sił traktują atomy jako kuleczki połączone sprężynami o stałych parametrach. Działają szybko i radzą sobie z dużymi białkami czy długimi skalami czasowymi, ale mają trudności ze subtelnymi zmianami kształtu, przestawianiem wiązań i ścieżkami reakcji istotnymi dla prawdziwej chemii. Metody kwantowe opisują natomiast elektrony wprost i potrafią dokładnie uchwycić rozrywanie i tworzenie wiązań. Są jednak tak wymagające obliczeniowo, że zwykle ograniczają się do małych cząsteczek lub bardzo krótkich symulacji. W ostatnich latach pojawiły się podejścia uczenia maszynowego jako środek pośredni, uczące się naśladować obliczenia kwantowe. Wiele z tych modeli albo brakuje fizycznej rygorystyczności potrzebnej do niezawodności, albo staje się zbyt wolne po rozszerzeniu na duże, realistyczne biomolekuły.
Nowy sposób uczenia SI o kształtach molekuł
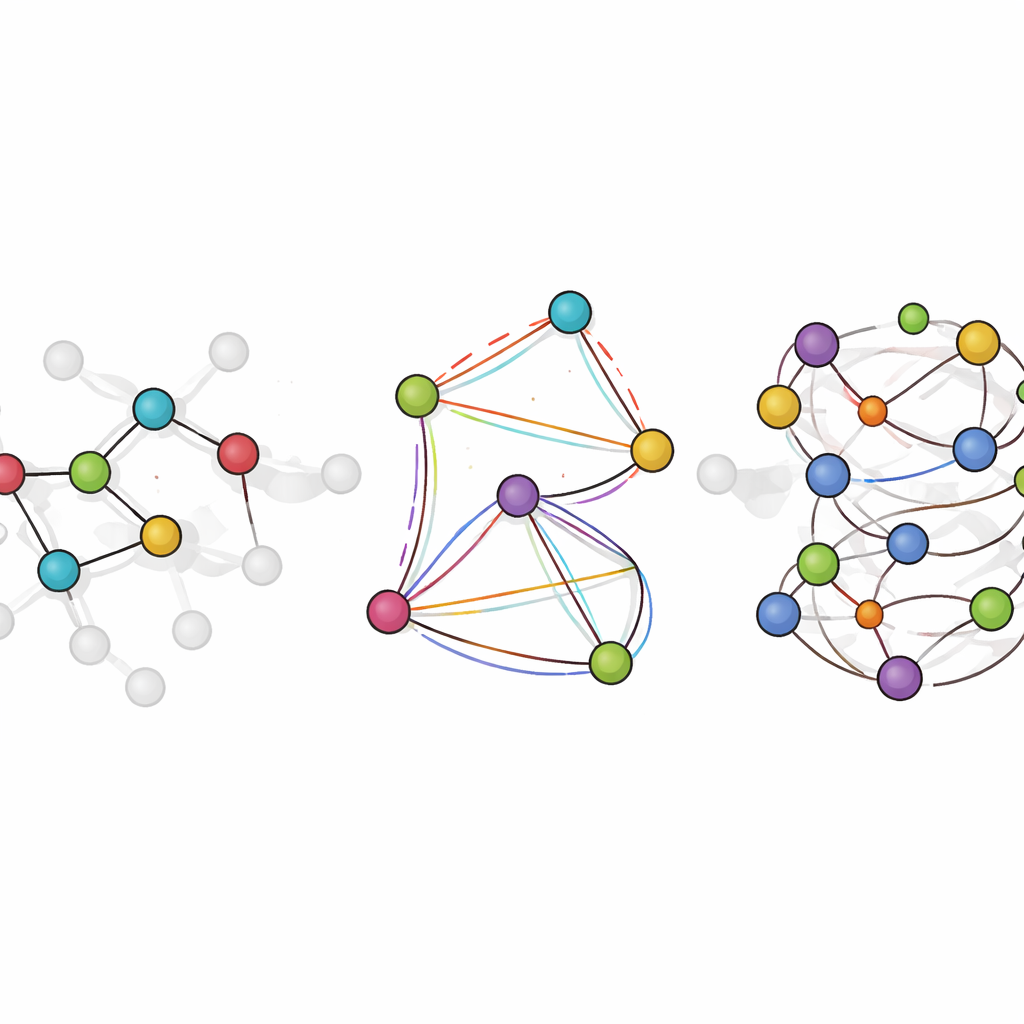
LiTEN rozwiązuje ten problem, przeprojektowując sposób, w jaki sieć neuronowa „odczuwa” geometrię molekuły. Zamiast uwzględniać jedynie proste odległości pomiędzy parami atomów, LiTEN wbudowuje informacje o wzorcach trój- i czteratomowych, które kontrolują kąty i skręty w cząsteczce. Co istotne, robi to z zachowaniem podstawowych symetrii fizycznych: jeśli obrócisz lub przesuniesz molekułę w przestrzeni, przewidywana energia pozostaje taka sama, a przewidywane siły obracają się odpowiednio. Kluczową innowacją, nazwaną tensoryzowaną uwagą czworokątną, jest możliwość uchwycenia złożonych interakcji zginania i skręcania przy użyciu efektywnych operacji wektorowych zamiast ciężkiej, wyspecjalizowanej matematyki. To utrzymuje obliczenia skalowalnymi, dzięki czemu wieloczłonowe efekty, które zwykle spowalniają zaawansowane modele, dają się obsłużyć przy koszcie rosnącym liniowo ze wzrostem rozmiaru układu.
Od uczenia na danych kwantowych do prawdziwych biomolekuł
Na bazie tej architektury autorzy zbudowali LiTEN-FF, „model podstawowy” dla sił molekularnych. Najpierw trenują go na masywnym zbiorze danych chemii kwantowej obejmującym miliony cząsteczek o cechach podobnych do leków, a następnie dopracowują na mniejszej, lecz dokładniejszej kolekcji zawierającej peptydy, struktury w roztworze i różnorodne pierwiastki, takie jak halogeny i metale. To dwuetapowe szkolenie pozwala modelowi nauczyć się zarówno szerokiego pokrycia chemicznego, jak i drobnych detali. W standardowych testach porównawczych, gdzie przewidywane energie i siły zestawia się z wynikami wysokopoziomowych obliczeń kwantowych, LiTEN dorównuje lub przewyższa wiodące modele zarówno dla małych, sztywnych cząsteczek, jak i znacznie większych, biologicznie istotnych układów. Utrzymuje dokładność nawet przy liczbie atomów sięgającej setek, zużywając przy tym mniej pamięci i działając szybciej niż wiele popularnych alternatyw.
Wykorzystanie modelu w symulowanej chemii
Ponad statyczne testy, zespół ocenia LiTEN-FF w zadaniach odzwierciedlających rzeczywiste przepływy pracy badawczej. Dla cząsteczek podobnych do leków potrafi optymalizować kształty tak, że otrzymane struktury niemal idealnie zgadzają się z wynikami wymagających obliczeń kwantowych, ale są szybsze o rząd wielkości. W symulacjach dynamiki molekularnej odtwarza fluktuacje długości wiązań i kątów w czasie, ściśle śledząc symulacje kwantowe. Doskonale przewiduje też zmiany energii przy obracaniu się cząsteczki wokół istotnego wiązania — kluczowy element uchwycenia preferencji konformacyjnych w projektowaniu leków. W wodzie ciekłej i w krótkich peptydach rozpuszczonych w wodzie LiTEN-FF generuje właściwości strukturalne i termodynamiczne zgodne zarówno z eksperymentami, jak i droższymi modelami referencyjnymi, oferując przy tym do dziesięciokrotnego przyspieszenia dla dużych układów.
Przyspieszanie poszukiwania użytecznych kształtów
Autorzy demonstrują też praktyczną procedurę wyszukiwania konformerów opartą na LiTEN-FF. Poprzez powtarzane cykle dynamiki w wysokiej temperaturze, chłodzenia i szybkiego udoskonalania geometrii, model generuje bogate zbiory odrębnych, niskoenergetycznych kształtów dla złożonych cząsteczek leków. W porównaniu z powszechnie stosowanym przepływem pracy opartym na obliczeniach kwantowych podejście napędzane SI znajduje bardziej zróżnicowane konformery w mniej niż połowie czasu. Co więcej, ponieważ LiTEN-FF potrafi przetwarzać wiele cząsteczek równolegle bez proporcjonalnego wzrostu kosztów, staje się szczególnie potężny w dużych kampaniach przesiewowych, gdzie trzeba ocenić tysiące kandydatów.
Co to oznacza dla przyszłości projektowania leków i materiałów
W istocie LiTEN-FF oferuje nowy silnik do symulacji molekularnych, który przybliża niezawodność na poziomie kwantowym do codziennej użyteczności. Dzięki bezpośredniemu kodowaniu geometrii kątów i skrętów w efektywnej sieci neuronowej zawęża przepaść między szybkimi, lecz przybliżonymi polami sił a wolnymi, lecz dokładnymi obliczeniami kwantowymi. Dla osób niebędących specjalistami kluczowy wniosek jest taki, że badacze wkrótce będą mogli uruchamiać znacznie bardziej realistyczne „eksperymenty” molekularne na komputerze, na skalę i z prędkościami zgodnymi ze współczesnym odkrywaniem leków i rozwojem materiałów. Przy szerokim przyjęciu i dalszym dopracowaniu modele z tej rodziny mogą stać się podstawowymi komponentami zautomatyzowanych pipeline’ów, które proponują, testują i dopracowują nowe molekuły na długo przed ich syntezą w laboratorium.
Cytowanie: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Słowa kluczowe: symulacja molekularna, pola sił uczone maszynowo, odkrywanie leków, modelowanie biomolekularne, chemia kwantowa