Clear Sky Science · pt
Um modelo base escalável e com precisão quântica para campos de força biomoleculares via atenção em quadriláteros tensorizada linearmente
Por que filmes moleculares mais rápidos importam
A descoberta de medicamentos moderna e o desenvolvimento de materiais dependem cada vez mais de “filmes” computacionais que mostram moléculas torcendo, flexionando e reagindo. Essas simulações podem revelar como um fármaco se encaixa em um bolso de proteína ou como um novo material se comporta sob tensão. Mas as ferramentas atuais forçam os cientistas a escolher: métodos rápidos que sacrificam precisão, ou cálculos quânticos ultra-precisos que são lentos demais para sistemas realistas e complexos. Este artigo apresenta o LiTEN e seu modelo de campo de força LiTEN-FF, uma nova abordagem de inteligência artificial que busca oferecer precisão ao nível quântico com velocidades adequadas para a modelagem molecular cotidiana.
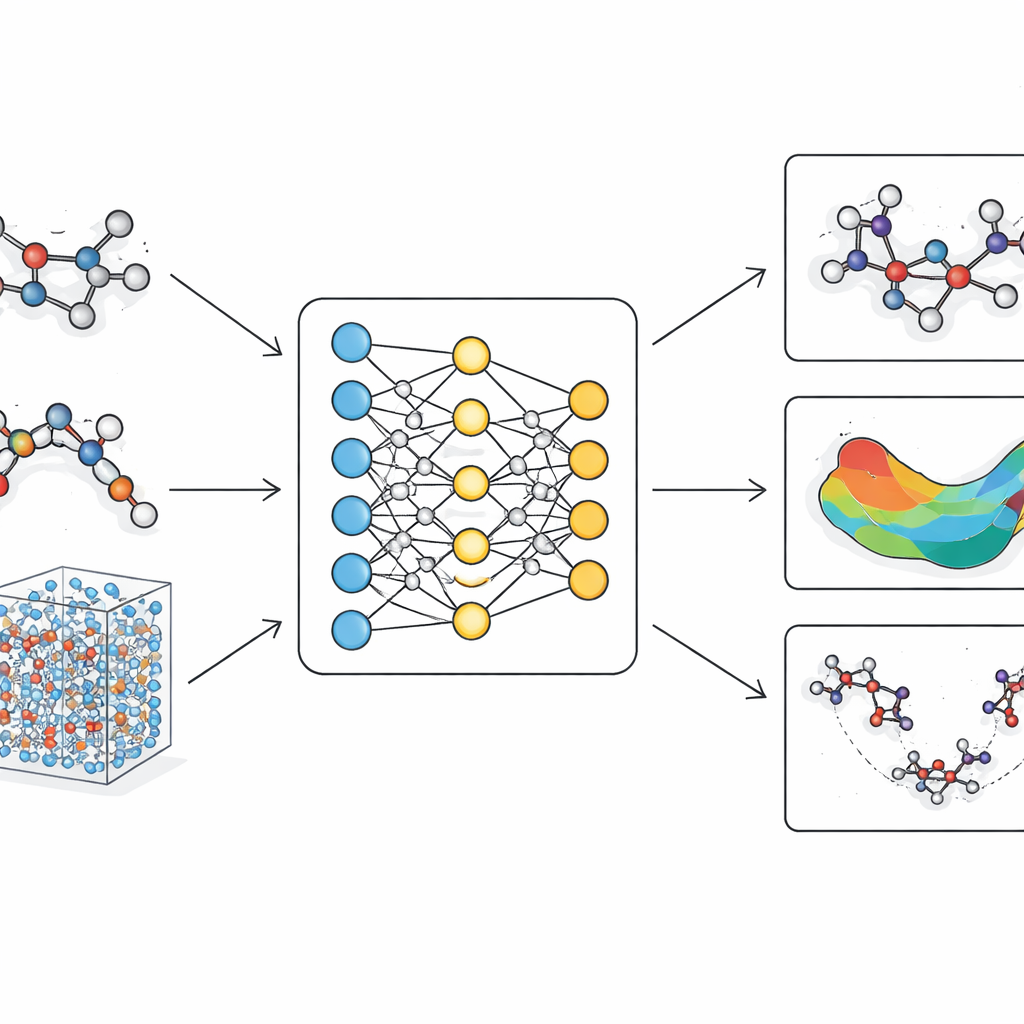
O problema dos modelos moleculares atuais
As simulações moleculares tradicionais dividem-se em dois campos. Campos de força clássicos tratam átomos como esferas conectadas por molas com parâmetros fixos. Eles são rápidos e conseguem lidar com proteínas grandes ou escalas de tempo longas, mas têm dificuldades com mudanças sutis de forma, rearranjos de ligações e caminhos de reação relevantes para a química real. Métodos quânticos, em contraste, descrevem explicitamente os elétrons e capturam com precisão a quebra e formação de ligações. Entretanto, são tão exigentes computacionalmente que costumam ficar limitados a moléculas pequenas ou a simulações muito curtas. Nos últimos anos, abordagens de aprendizado de máquina surgiram como um caminho intermediário, aprendendo a imitar cálculos quânticos. Ainda assim, muitos desses modelos carecem do rigor físico necessário para confiabilidade ou tornam-se lentos demais ao serem escalados para biomoléculas grandes e realistas.
Uma nova forma de ensinar IA sobre formas moleculares
O LiTEN enfrenta esse desafio redesenhando a forma como uma rede neural “sente” a geometria molecular. Em vez de considerar apenas distâncias pareadas simples entre átomos, o LiTEN incorpora informações sobre padrões de três e quatro átomos que controlam ângulos e torções na molécula. Crucialmente, faz isso respeitando simetrias físicas básicas: se você rotacionar ou mover uma molécula no espaço, sua energia prevista permanece a mesma, e as forças previstas rotacionam de modo adequado. A inovação-chave, chamada atenção em quadriláteros tensorizada, permite que o modelo capture interações complexas de flexão e torção usando operações vetoriais eficientes em vez de matemática pesada e especializada. Isso mantém o cálculo escalável, de modo que efeitos de muitos corpos que normalmente sobrecarregam modelos avançados podem ser tratados com um custo que cresce apenas linearmente com o tamanho do sistema.
Do treinamento em dados quânticos a biomoléculas reais
Sobre essa arquitetura, os autores constroem o LiTEN-FF, um “modelo base” para forças moleculares. Eles o treinam primeiro em um enorme conjunto de dados de química quântica com milhões de moléculas semelhantes a fármacos, depois o refinam em uma coleção menor, porém de maior precisão, que inclui peptídeos, estruturas solvatas e elementos diversos como halogênios e metais. Esse treinamento em duas etapas permite que o modelo aprenda tanto uma cobertura química ampla quanto detalhes finos. Em testes-padrão que comparam energias e forças previstas com resultados quânticos de alto nível, o LiTEN iguala ou supera os modelos líderes em moléculas pequenas e rígidas e em sistemas biológicos muito maiores. Mantém a precisão mesmo quando o número de átomos cresce para centenas, usando menos memória e rodando mais rápido do que muitas alternativas populares.
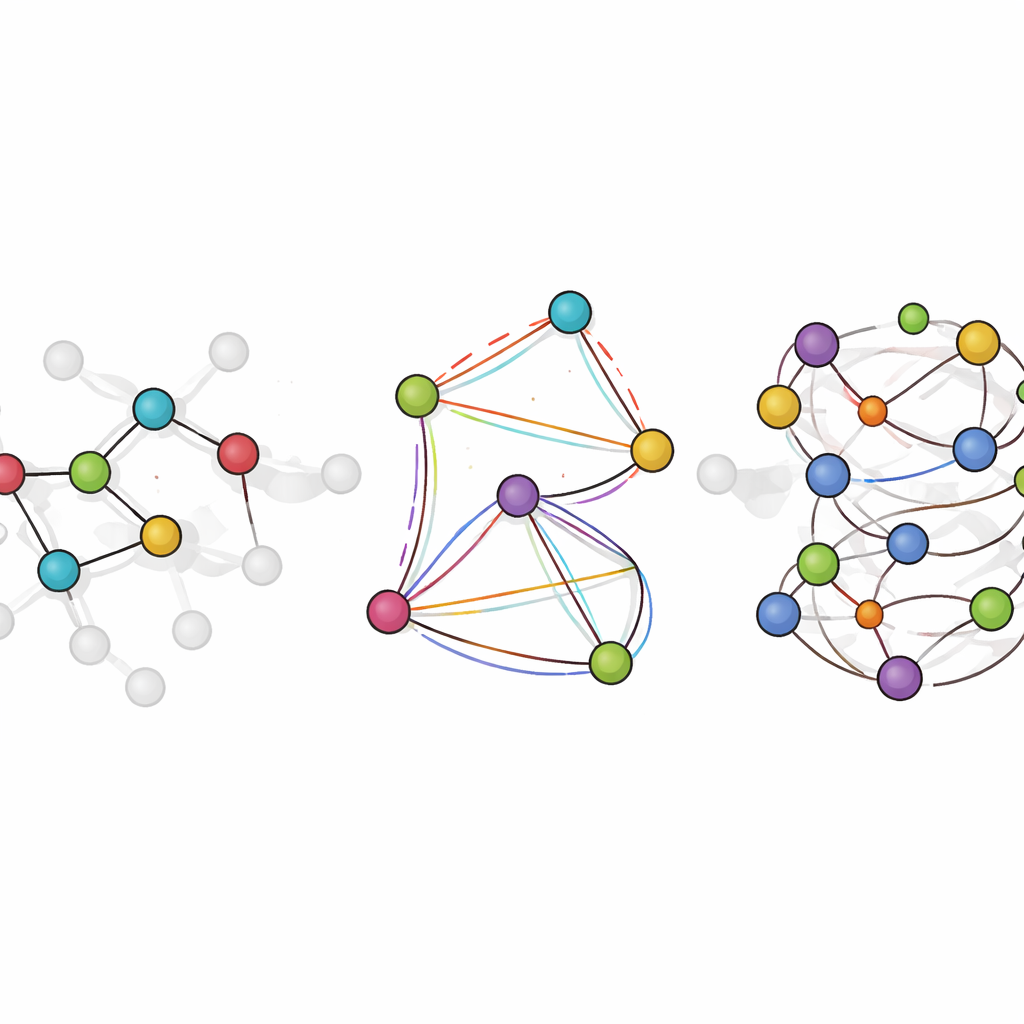
Colocando o modelo para funcionar na química simulada
Além de testes estáticos, a equipe avalia o LiTEN-FF em tarefas que espelham fluxos de trabalho de pesquisa reais. Para moléculas semelhantes a fármacos, ele pode otimizar conformações de modo que as estruturas resultantes quase coincidem com as obtidas por cálculos quânticos exigentes, mas milhares de vezes mais rápido. Em simulações de dinâmica molecular, reproduz como comprimentos de ligação e ângulos flutuam ao longo do tempo, acompanhando de perto simulações baseadas em métodos quânticos. Também se destaca ao prever como a energia muda quando uma molécula gira em torno de uma ligação chave, um ingrediente crucial para capturar preferências conformacionais no projeto de fármacos. Em água líquida e em peptídeos curtos dissolvidos em água, o LiTEN-FF gera propriedades estruturais e termodinâmicas que concordam bem com experimentos e com modelos de referência mais caros, ao mesmo tempo em que oferece acelerações de até dez vezes em sistemas grandes.
Acelerando a busca por formas úteis
Os autores também demonstram um pipeline prático de busca de conformadores construído em torno do LiTEN-FF. Executando ciclos repetidos de dinâmica em alta temperatura, resfriamento e refinamento geométrico rápido, o modelo gera conjuntos ricos de formas distintas e de baixa energia para moléculas farmacêuticas complexas. Em comparação com um fluxo de trabalho largamente usado baseado em métodos quânticos, essa abordagem dirigida por IA encontra conformadores mais diversos em menos da metade do tempo. Além disso, porque o LiTEN-FF pode processar muitas moléculas em paralelo sem aumento proporcional de custo, torna-se particularmente poderoso para campanhas de triagem em larga escala, onde milhares de candidatos precisam ser avaliados.
O que isso significa para o futuro do projeto de fármacos e materiais
Em essência, o LiTEN-FF oferece um novo motor para simulação molecular que aproxima muito mais a confiabilidade ao nível quântico da usabilidade cotidiana. Ao codificar a geometria de ângulos e torções diretamente em uma rede neural eficiente, ele reduz a lacuna entre campos de força rápidos, mas aproximados, e cálculos quânticos lentos, porém precisos. Para não especialistas, a conclusão é que os pesquisadores em breve poderão executar “experimentos” moleculares muito mais realistas no computador, em escalas e velocidades compatíveis com a descoberta de medicamentos e o desenvolvimento de materiais modernos. Se amplamente adotados e ainda refinados, modelos dessa família poderão se tornar componentes centrais de pipelines automatizados que propõem, testam e refinam novas moléculas muito antes de serem sintetizadas em laboratório.
Citação: Su, Q., Zhu, K., Gou, Q. et al. A scalable and quantum-accurate foundation model for biomolecular force fields via linearly tensorized quadrangle attention. Nat Commun 17, 3639 (2026). https://doi.org/10.1038/s41467-026-70377-4
Palavras-chave: simulação molecular, campos de força por aprendizado de máquina, descoberta de medicamentos, modelagem biomolecular, química quântica